Tumor de Wilms
Incidencia del tumor de Wilms
El tumor de Wilms es el tumor renal más frecuente durante la lactancia y la niñez. La incidencia del tumor de Wilms es de 10,4 casos por millón de niños menores de 15 años y de 0,2 casos por cada 10 000 lactantes.[1,2] En los Estados Unidos, cada año se diagnostican alrededor de 650 casos de tumor de Wilms. La incidencia es mucho más baja en las personas asiáticas.[1,3]
La proporción hombre a mujer de casos unilaterales de tumor de Wilms es de 0,92:1,00, pero en los casos bilaterales se presentan con mayor frecuencia en las mujeres (0,60). La media de edad en el momento del diagnóstico es de 44 meses en los casos unilaterales y de 31 meses en los casos bilaterales de tumor de Wilms.[4,5] Cerca del 10 % de los niños con tumor de Wilms tienen un síndrome de malformación congénita relacionado.[6]
Síndromes y otras afecciones relacionadas con el tumor de Wilms
Por lo general, el tumor de Wilms se presenta en niños sanos sin predisposición al cáncer. Sin embargo, se notificó que cerca del 10 % de los niños con tumor de Wilms presentan una anomalía congénita.[6,7] En pacientes con anomalías congénitas y tumor de Wilms, se notificaron restos nefrogénicos en el 60 % de los casos.[8] De 295 pacientes consecutivos con tumor de Wilms atendidos en el Institut Curie de París, 52 (17,6 %) presentaron anomalías o síndromes; entre ellos, 43 se consideraron mayores y de estos, 14 fueron síndromes de predisposición tumoral comprobados mediante análisis genético.[9]
Los niños con tumor de Wilms pueden presentar hemihipertrofia y anomalías relacionadas en el aparato urinario, incluso criptorquidia e hipospadias. Es posible que los niños exhiban síndromes fenotípicos reconocibles, como sobrecrecimiento, aniridia y anomalías genéticas, entre otros. Estos síndromes han proporcionado pistas sobre el origen genético de la enfermedad. Los síndromes fenotípicos y otras afecciones se agrupan en categorías de sobrecrecimiento y ausencia de sobrecrecimiento (consultar el Cuadro 1). Los síndromes y las afecciones de sobrecrecimiento se caracterizan por crecimiento somático prenatal y posnatal excesivo.[10,11]
Es importante reconocer que el riesgo absoluto de tumor de Wilms varía de acuerdo con la afección o anomalía subyacentes. Por ejemplo, la mayoría de los pacientes con hemihipertrofia no presentarán tumor de Wilms.
| Síndrome o afección | Gen | Fenotipo de sobrecrecimiento | Fenotipo sin sobrecrecimiento |
|---|---|---|---|
| Riesgo alto de tumor de Wilms (>20 %) | |||
| CLOVES = sobrecrecimiento lipomatoso congénito, malformaciones vasculares, nevos epidérmicos y anomalías esqueléticas o vertebrales; MULIBREY (muscles, liver, bran, eyes) = anomalías características en los músculos, el hígado, el encéfalo y los ojos; WAGR = tumor de Wilms, aniridia, anomalía genitourinaria y una variedad de alteraciones en el desarrollo neurológico. | |||
| a Adaptación de Treger et al.[12] | |||
| Síndrome WAGR (espectro WAGR) | Deleción deWT1: | X | |
| Síndrome de Denys-Drash | Variante de aminoácido enWT1 | X | |
| Síndrome de Perlman | Variante deDIS3L2 | X | |
| Anemia de Fanconi con variantes bialélicas deBRCA2(FANCD1) oPALB2(FANCN) | BRCA2,PALB2 | X | |
| Separación prematura de la cromátida y aneuploidia en mosaico variegada | Variante bialélica deBUB1BoTRIP13 | X | |
| Riesgo moderado de tumor de Wilms (5–20 %) | |||
| Síndrome de Frasier | Variante en el sitio de empalme en el intrón 9 deWT1 | X | |
| Síndrome de Beckwith-Wiedemann | Disomía uniparental o epivariante de H19 | X | |
| Síndrome de Simpson-Golabi-Behmel | Variante deGPC3 | X | |
| Riesgo bajo de tumor de Wilms (<5 %) | |||
| Síndrome de Bloom | Variante bialélica deBLM | X | |
| Síndrome DICER1 | Variante deDICER1 | X | |
| Síndrome de Li-Fraumeni | TP53,CHEK2 | X | |
| Hemihipertrofia aislada | X | ||
| Hiperparatiroidismo-síndrome de tumor de mandíbula | Variante deCDC73(también conocido comoHRPT2) | X | |
| Síndrome de enanismo MULIBREY | Variante deTRIM37 | X | |
| Sobrecrecimiento segmentario relacionado conPIK3CAque incluye el síndrome CLOVES | Variante dePIK3CA | X | |
| Síndrome de microdeleción de 9q22.3 | 9q22.3 | X | |
| Síndrome de Sotos | NSD1 | X | |
| Tumor de Wilms familiar | FWT1 | X | |
| FWT2 | |||
| Anomalías genitourinarias | WT1 | X | |
| Aniridia esporádica | WT1 | X | |
| Trisomía 18 | X | ||
Para obtener información sobre los genes relacionados con el tumor de Wilms, incluso WT1 y WT2, consultar la sección Características genómicas del tumor de Wilms.
Causas sindrómicas del tumor de Wilms
Síndromes relacionados con el genWT1
Los síndromes relacionados con el gen WT1 son los siguientes:
- Síndrome WAGR (espectro WAGR).[13] El síndrome WAGR se caracteriza por los siguientes aspectos:
- Tumor de Wilms (W ilms tumor)
- Aniridia (A niridia).
- Anomalía genitourinaria (G enitourinary anomaly)
- Alteraciones en el desarrollo neurológico (R ange of developmental delays).
La constelación del síndrome WAGR se asocia con una deleción intersticial en el cromosoma 11 (del(11p13)). La prevalencia de esta deleción es de alrededor del 0,4 % de los niños con tumor de Wilms.[14,15] El riesgo de que se presente tumor de Wilms en niños con síndrome WAGR es de alrededor del 50 %. Estos niños presentarán tumores de Wilms más temprano (mediana de edad, 22 meses) y tendrán una incidencia más alta de tumores bilaterales (37 %) que los niños con tumores de Wilms no sindrómicos.[16,17] De 43 pacientes con síndrome WAGR que presentaron tumor de Wilms o nefroblastomatosis, ninguno presentó metástasis ni exhibió características histológicas anaplásicas. De los pacientes, 3 presentaron tumores contralaterales, uno de los cuales se presentó 7 años después del diagnóstico inicial.[17] Para obtener más información, consultar la sección Características genómicas del tumor de Wilms.
- Síndrome de Denys-Drash y síndrome de Frasier. Las anomalías genitourinarias, como el hipospadias, la criptorquidia y otras, se relacionan con variantes de WT1 (prevalencia de alrededor del 8 % al 10 % en los niños con tumor de Wilms). Los niños con genoma XY que tienen seudohermafroditismo o enfermedad renal (glomerulonefritis o síndrome nefrótico) y presentan tumor de Wilms a veces tienen un síndrome de Denys-Drash o Frasier (caracterizado por hermafroditismo masculino, amenorrea primaria, insuficiencia renal crónica y otras anomalías).[18] Ambos síndromes se asocian con variantes del gen WT1.[19] En concreto, las variantes germinales de aminoácido del gen WT1 son responsables de la mayoría de los casos de tumor de Wilms que se presentan como parte del síndrome de Denys-Drash.[20,21] El riesgo de tumor de Wilms es de alrededor del 90 % para los niños con síndrome de Denys-Drash y la enfermedad bilateral se presenta en el 20 % de los pacientes.[21,22] En el síndrome de Frasier, las variantes en el sitio de empalme de WT1 producen un desequilibrio de las isoformas de WT1 y una incidencia mucho más baja del tumor de Wilms.[23]
Síndromes relacionados con el genWT2
Los síndromes relacionados con el gen WT2 son los siguientes:
- Síndrome de Beckwith-Wiedemann. El síndrome de Beckwith-Wiedemann es un síndrome de sobrecrecimiento que se caracteriza por un crecimiento asimétrico de una o más partes del cuerpo, lengua grande, onfalocele o hernia umbilical al nacer, pliegues u hoyos en la piel cerca de las orejas, anomalías renales e hipoglucemia (en neonatos). En un estudio de vinculación de registros poblacionales de todos los nacidos vivos en Texas entre 1999 y 2017, los niños con síndrome de Beckwith-Wiedemann tenían una probabilidad 42 veces mayor de presentar cáncer pediátrico. El hepatoblastoma fue el cáncer más común, seguido del tumor de Wilms. El porcentaje de niños con síndrome de Beckwith-Wiedemann diagnosticados de cáncer fue del 1,24 % a los 5 años de edad, del 5,58 % a los 10 años y del 10,81 % a los 15 años. La presencia de cualquier característica aislada de sobrecrecimiento se relacionó con el riesgo de presentar cáncer (cociente de riesgos instantáneos [CRI], 4,70). La hepatoesplenomegalia (CRI, 23,04) y la macroglosia (CRI, 11,18) se relacionaron con el riesgo de cáncer de forma más sólida.[24] Alrededor del 15 % de los niños con síndrome de Beckwith-Wiedemann tendrán tumores bilaterales.[25]
El síndrome de Beckwith-Wiedemann obedece a una alteración en la expresión de dos complejos génicos que participan en el control del crecimiento y el avance del ciclo celular mediante la regulación de dos regiones independientes de control de impronta (ICR1 [ICR telomérica] e ICR2 [ICR centromérica]) en el cromosoma 11p15.5. Estas dos ICR se caracterizan por metilación diferencial de los alelos maternos y paternos. En la etiopatogenia del síndrome de Beckwith-Wiedemann participan varios mecanismos moleculares que producen expresión desequilibrada de genes de impronta en los dos dominios mencionados. La predisposición tumoral se produce ante todo por la desregulación del dominio telomérico de 11p15 (ganancia de metilación en ICR1 [ICR1-GoM] y disomía uniparental paterna [DUP]) más que en el dominio centromérico de 11p15 (pérdida de metilación en ICR2 [ICR2-LoM] y una variante de CDKN1C).[26] Cerca del 15 % de los casos con fenotipos bien definidos no tienen defectos moleculares establecidos hasta el momento.[27,28]
Los subtipos moleculares del síndrome predisponen a los pacientes a presentar diferentes histotipos tumorales.[29,30,31]
La prevalencia del síndrome de Beckwith-Wiedemann se notificó antes como del 1 % en los niños con tumor de Wilms.[25,32,33,34] Sin embargo, en un estudio nacional holandés de cohortes seguidas durante 5 años, se demostró que el 16 % de los pacientes con tumor de Wilms (20 de 126) tenían síndrome de Beckwith-Wiedemann. En este estudio se incluyeron tanto pacientes con diagnóstico clínico como pacientes en los que no se observó el fenotipo del síndrome de Beckwith-Wiedemann (ganancia de metilación de 11p15 ICR1 en parénquima renal y sangre periférica normales). Es probable que el mosaicismo explique los casos de fenotipo oculto.[35] En conjunto, cerca del 10 % de los pacientes con síndrome de Beckwith-Wiedemann presentarán tumor de Wilms. Sin embargo, esta incidencia varía según el epigenotipo. Los niños con ICR1-GoM tienen el riesgo más alto de presentar tumor de Wilms (22–29 %). Los niños con DUP paterna tienen un riesgo más bajo (7–17 %) y los pacientes con ICR2-LoM y variantes de CDKN1C tienen un riesgo mínimo.[26,30,31] Los pacientes con síndrome de Beckwith-Wiedemann y hemihipertrofia tienen 4 veces más riesgo de tumor que los pacientes con síndrome de Beckwith-Wiedemann sin hemihipertrofia.[36] Para obtener más información, consultar la sección Características genómicas del tumor de Wilms.
Otras causas sindrómicas del tumor de Wilms
Otras causas sindrómicas del tumor de Wilms son las siguientes:
- Síndrome de Perlman. Se trata de un síndrome de sobrecrecimiento congénito de herencia autosómica recesiva poco frecuente. Se caracteriza por gigantismo fetal, displasia renal y nefroblastomatosis, hipertrofia de las células de los islotes, múltiples anomalías congénitas y discapacidad intelectual. Los sobrevivientes tienen un riesgo alto de presentar tumor de Wilms (75 %).[37]
Las variantes inactivadoras germinales de DIS3L2 en el cromosoma 2q37 se relacionan con el síndrome de Perlman. Los datos preliminares indican que DIS3L2 desempeña una función en el desarrollo normal de los riñones y en un subconjunto de casos esporádicos de tumor de Wilms.[38]
Las variantes constitucionales heterocigotas de DIS3L2 parecen tener una relación con la predisposición al tumor de Wilms. En un estudio nacional holandés de cohortes seguidas durante 5 años, el 4 % de los pacientes con tumores de Wilms (5 de 126) tenían variantes de DIS3L2. Sin embargo, es probable que la penetrancia real sea mucho más baja que en los casos homocigóticos (síndrome de Perlman).[35]
- Síndrome de Simpson-Golabi-Behmel. Este síndrome se caracteriza por macroglosia, macrosomía, anomalías renales y esqueléticas, y mayor riesgo de cánceres embrionarios.
El síndrome es causado por variantes o deleciones de los genes GPC3 y GPC4; se cree que estas anomalías genéticas aumentan el riesgo de tumor de Wilms (8 %).[39]
- Síndrome CLOVES. Este síndrome se caracteriza por los siguientes aspectos:
- Sobrecrecimiento lipomatoso congénito (C ongenital L ipomatous O vergrowth).
- Defectos vasculares (V ascular malformations).
- Nevos epidérmicos (E pidermal nevi).
- Anomalías esqueléticas o vertebrales (S keletal/spinal abnormalities).
La causa de este síndrome son variantes somáticas poscigóticas de PIK3CA, que afectan partes del cuerpo grandes o pequeñas del niño.[40]
- Síndrome de Sotos. El síndrome de Sotos se caracteriza por gigantismo cerebral y dificultades de aprendizaje que oscilan de leves a graves. Este síndrome se relaciona con problemas de comportamiento, anomalías cardíacas congénitas, ictericia neonatal, escoliosis, convulsiones y anomalías renales, como tumor de Wilms.
Las variantes del gen NSD1 son la única causa conocida del síndrome de Sotos.[41]
- Síndrome de microdeleción de 9q22.3. Este síndrome se caracteriza por anomalías craneofaciales, craneosinostosis metópica, hidrocefalia, macrosomía y dificultades de aprendizaje.
De los 44 pacientes descritos con deleciones de 9q22.3, 7 presentaron tumor de Wilms, y hubo una asociación con sobrecrecimiento en 4 de esos 7 pacientes. Aunque el tamaño de las deleciones fue variable, todas ellas abarcaron el gen PTCH1.[42]; [43][Nivel de evidencia C1] Según los autores de este estudio, se debe considerar la vigilancia para el tumor de Wilms en cualquier paciente con síndrome de microdeleción de 9q22.3, en especial si tiene sobrecrecimiento.[43][Nivel de evidencia C1]
- Síndrome de Bloom. Este síndrome se caracteriza por estatura baja y más delgadez que otros miembros de la familia, cambios en la piel por sensibilidad al sol y un mayor riesgo de tumor de Wilms.
Las variantes del gen BLM son la única causa conocida del síndrome de Bloom.[44]
- Síndrome de Li-Fraumeni. Este síndrome es un trastorno raro que aumenta mucho el riesgo de varios tipos de cáncer, en especial durante la niñez y la juventud. Los tipos de cáncer que se relacionan más a menudo con el síndrome de Li-Fraumeni son el cáncer de mama, el osteosarcoma, el sarcoma de tejido blando, los tumores de encéfalo, la leucemia, el carcinoma de corteza suprarrenal y el tumor de Wilms.
Se encuentra una variante del gen TP53 en la mayoría de las familias con síndrome de Li-Fraumeni. También se sabe que la variante del gen CHEK2 causa el síndrome de Li-Fraumeni.[45]
- Síndrome de Alagille. Este síndrome incluye cardiopatía congénita, deformidades faciales y anormalidades vertebrales, oculares y renales. Se notificó junto con el tumor de Wilms en dos pacientes con variantes identificadas.[46]
- Síndrome de Bohring-Opitz. Este síndrome es una afección genética rara caracterizada por rasgos faciales diferenciados, microcefalia variable, hipertricosis, nevo flámeo, miopía grave, postura extraña, discapacidad intelectual grave y problemas de alimentación.
El síndrome se relaciona con variantes de ASXL1 y una incidencia estimada de tumor de Wilms del 7 %.[47]
Causas no sindrómicas del tumor de Wilms
Las causas no sindrómicas del tumor de Wilms son las siguientes:
- Tumor de Wilms familiar. A pesar del número de genes que participan en la formación del tumor de Wilms, el tumor de Wilms familiar es poco frecuente; cerca del 2 % de los pacientes tiene antecedentes familiares de tumor de Wilms. Los hermanos o hermanas de ambos sexos de los pacientes con tumor de Wilms tienen una probabilidad de presentar la enfermedad menor al 1 %.[48,49,50] El riesgo de tumor de Wilms en la descendencia de las personas que han tenido tumores unilaterales (esporádicos) es menor al 2 %.[51]
Se identificaron dos locus de distribución en 17q12-q21 (FWT1) y 19q13.4 (FWT2) mediante estudios de ligamiento genético de familias afectadas por tumor de Wilms. Aunque los genes aún no se han caracterizado, en hermanos de ambos sexos con tumor de Wilms se detectó pérdida de función del correpresor transcripcional TRIM28, que se encuentra en FWT2.[52,53,54] En ocasiones, hay familias con tumor de Wilms que tienen variantes germinales de WT1. En estas familias, la mayoría de los miembros, pero no todos, tienen malformaciones del aparato genitourinario.[55,56]
Se encontraron variantes inactivadoras de CTR9 en 3 de 35 familias con tumor de Wilms. El gen CTR9 que se ubica en 11p15.3 es un componente fundamental del complejo del factor asociado con la polimerasa 1 (PAF1), que tiene múltiples funciones en la regulación de la ARN–polimerasa II y en la elongación transcripcional, además participa en la organogénesis embrionaria.[57] Algunas familias con tumor de Wilms familiar tienen variantes germinales de microdeleción o microinserción en la región H19 de 11p15.3 que producen hipermetilación del sitio.[58]
- Anomalías constitucionales de 11p15. Se encontraron anomalías constitucionales de 11p15 en el DNA linfocitario de 13 entre 437 personas (3 %) con tumor de Wilms esporádico sin manifestaciones de trastornos del crecimiento, entre estos, 12 % de los casos eran bilaterales. Todas las anomalías eran nuevas y poscigóticas, excepto una microdeleción nueva en un niño cuya madre tenía la variante pero no estaba afectada; sin embargo, un hermano menor que presentaba la microdeleción tenía síndrome de Beckwith-Wiedemann. Esto indica que se debe considerar el análisis constitucional de 11p15 en todas las personas con tumor de Wilms.[58]
- Aniridia esporádica. La aniridia esporádica a veces se produce por deleciones germinales pequeñas en una copia del gen PAX6, que afecta una parte o la totalidad del gen WT1 adyacente, pero que no produce anomalías genitourinarias ni discapacidad intelectual (es decir, sin un síndrome WAGR obvio). En consecuencia, muchos pacientes con aniridia esporádica que presentan tumor de Wilms son aptos para hacerse pruebas genéticas. El riesgo relativo de un tumor de Wilms en pacientes con aniridia esporádica es 67 veces más alto.[59] Alrededor de la mitad de las personas con aniridia esporádica y deleciones de PAX6 y WT1 presentan tumor de Wilms.[60]
- Hemihipertrofia aislada (también conocida como sobrecrecimiento lateralizado o hemihiperplasia). La hemihipertrofia es un sobrecrecimiento asimétrico de una o más partes del cuerpo en ausencia de un patrón reconocido de malformaciones, displasia o variantes morfológicas y se ha relacionado con el tumor de Wilms.[61] También se puede relacionar con otros síndromes de predisposición, como el síndrome de Beckwith-Wiedemann. Es posible que los signos clínicos no sean muy evidentes y se puede observar hemihipertrofia después del diagnóstico de un tumor.
En los pacientes con hemihipertrofia aislada e isodisomía uniparental paterna de 11p15.5, se estima que el riesgo de tumor de Wilms es de alrededor del 8 %.[62]
- Trisomía 18.[63]
- Anemia de Fanconi con variantes bialélicas de BRCA2 (FANCD1) o PALB2 (FANCN). Los genes BRCA2 y PALB2 desempeñan una función central en la reparación de la recombinación homóloga del DNA. Las variantes bialélicas de BRCA2 o PALB2 conducen a anemia de Fanconi y a un aumento del riesgo de ciertos cánceres infantiles, como el tumor de Wilms.[64,65,66]
- Exposición materna a plaguicidas. En un estudio de población francés, el uso materno de cualquier pesticida doméstico durante el embarazo se relacionó con un riesgo de tumor de Wilms en niños (oportunidad relativa [OR], 1,6). Los insecticidas fueron el tipo de plaguicida que se notificó con mayor frecuencia, y la relación con el tumor de Wilms fue más fuerte cuando los insecticidas se usaron más de una vez al mes.[67][Nivel de evidencia C1]
Características genómicas del tumor de Wilms
Características moleculares del tumor de Wilms
El tumor de Wilms a veces surge durante la embriogénesis en el contexto de un riñón que por lo demás es normal desde el punto de vista genómico; en otras ocasiones, surge de lesiones precursoras genéticas somáticas no germinales presentes dentro de un tejido renal con características histológicas y funcionales normales. La hipermetilación de H19, un componente conocido de un subconjunto de tumores de Wilms, es una anomalía genética muy común que se encuentra en estas áreas de apariencia normal de las lesiones precursoras.[68]
En un estudio se obtuvo la secuenciación del genoma completo, la expresión de mRNA y miRNA, el número de copias de DNA y el análisis de metilación de117 tumores de Wilms; luego se hizo la secuenciación dirigida de 651 tumores de Wilms.[69] Se seleccionaron tumores de Wilms recidivantes con características histológicas favorables (HF) o tumores que exhibían anaplasia difusa. En el estudio se observaron las siguientes características:[69]
- Los tumores de Wilms surgen a menudo como consecuencia de más de una alteración genética.
- Los tumores de Wilms con anomalías genéticas diferentes exhiben diferencias en la expresión génica y los patrones de metilación.
- En los tumores de Wilms hay un gran número de genes iniciadores propuestos como candidatos, y casi todos estos están alterados en menos del 5 % de los tumores de Wilms.
- Los tumores de Wilms exhiben variantes recurrentes de genes con funciones comunes; la mayoría de ellos participan en el desarrollo renal temprano o en la regulación epigenética (por ejemplo, modificaciones de la cromatina, elongación transcripcional y miRNA).
Alrededor de un tercio de los casos de tumor de Wilms tienen variantes de los genes WT1, CTNNB1 o AMER1 (WTX).[70,71] Otro subgrupo de casos de tumor de Wilms surgen por variantes de los genes procesadores de miRNA (miRNAPG), como DROSHA, DGCR8, DICER1 y XPO5.[72,73,74,75] También se observan alteraciones recurrentes en otros genes fundamentales durante el desarrollo renal temprano como SIX1 y SIX2 (factores de transcripción que cumplen una función clave en el desarrollo renal temprano),[72,73]EP300, CREBBP y MYCN.[69] De las variantes que se encuentran en los tumores de Wilms, el 30 % al 50 % convergen en el proceso de elongación transcripcional durante el desarrollo renal, estas variantes afectan los genes MLLT1, BCOR, MAP3K4, BRD7 y HDAC4.[69] El tumor de Wilms anaplásico se caracteriza por presentar variantes de TP53.
Se observan tasas altas de tumor de Wilms en pacientes con una variedad de trastornos genéticos, como el síndrome WAGR (tumor de Wilms, aniridia, anormalidades genitourinarias y una variedad de alteraciones en el desarrollo neurológico), el síndrome de Beckwith-Wiedemann, la hemihipertrofia, el síndrome de Denys-Drash y el síndrome de Perlman.[76] Se han identificado otras causas genéticas en casos de tumor de Wilms familiar, como las variantes germinales de REST y CTR9.[57,77]
A continuación se resumen las características genómicas y genéticas del tumor de Wilms.
GenWT1
El gen WT1 se encuentra en el brazo corto del cromosoma 11 (11p13). WT1 es un factor de transcripción necesario para el desarrollo genitourinario normal y es fundamental para la diferenciación del blastema renal.[78] Las variantes de WT1 se observan en el 10 % al 20 % de los casos de tumor de Wilms esporádico.[70,78,79]
El tumor de Wilms con variante de WT1 se caracteriza por los siguientes aspectos:
- A menudo presenta activación de la vía WNT por variantes activadoras del gen CTNNB1.[79,80,81]
- Frecuentemente se observa la pérdida de heterocigosidad (LOH) en 11p15, debido a que la disomía uniparental paterna en el cromosoma 11 representa un mecanismo común de pérdida del alelo de WT1 normal remanente.[79,82]
- Los restos nefrogénicos son centros benignos de células renales embrionarias que persisten de manera anormal durante el periodo posnatal. Se encuentran restos nefrogénicos intralobulares en casi el 20 % de los casos de tumor de Wilms. Estos restos se observan con bastante frecuencia en los casos de síndromes genéticos que tienen variantes de WT1, como los síndromes WAGR y de Denys-Drash.[83] También se observan restos nefrogénicos intralobulares en casos con variantes esporádicas de los genes WT1 y MLLT1.[84,85]
- Las variantes germinales de WT1 son infrecuentes (2–4 %) en el tumor de Wilms no sindrómico.[56,86]
- En un estudio de 56 pacientes que no habían recibido quimioterapia, las variantes de WT1 y la LOH de 11p15 se relacionaron con recidiva en pacientes con tumores de Wilms de riesgo muy bajo.[87] Estos resultados requieren validación y quizás proporcionen biomarcadores para la clasificación de pacientes en el futuro.
Las variantes germinales de WT1 son más comunes en los niños que tienen tumor de Wilms y presentan una de las siguientes afecciones:
- Síndrome WAGR, síndrome de Denys-Drash [21] o síndrome de Frasier.[18]
- Anomalías genitourinarias, incluso hipospadias y criptorquidia.
- Tumor de Wilms bilateral.
- Tumor de Wilms unilateral con restos nefrogénicos en el riñón contralateral.
- Diferenciación estromal y rabdomiomatosa.
Las variantes germinales de un solo nucleótido de WT1 producen síndromes genéticos caracterizados por nefropatía, trastorno del desarrollo sexual 46XY y riesgos variables de tumor de Wilms.[88,89] Las afecciones sindrómicas con variantes germinales de WT1, incluyen el síndrome WAGR, el síndrome de Denys-Drash [21] y el síndrome de Frasier.[18]
- Síndrome WAGR. Los niños con síndrome WAGR tienen un riesgo alto (cerca del 50 %) de presentar tumor de Wilms.[6] El síndrome WAGR se produce por deleciones en el cromosoma 11p13 que afectan un conjunto de genes contiguos, entre ellos, los genes WT1 y PAX6.
Las variantes inactivadoras o las deleciones del gen PAX6 producen aniridia, mientras que la deleción de WT1 aumenta el riesgo de tumor de Wilms. La pérdida del gen LMO2 se relacionó con una aparición más frecuente de tumor de Wilms en pacientes con aniridia congénita y deleciones en la región WAGR.[90][Nivel de evidencia C1] La aniridia esporádica sin deleción de WT1 no se relaciona con aumento de riesgo de tumor de Wilms. En consecuencia, los niños con aniridia familiar (que por lo general afecta a muchas generaciones), pero sin anomalías renales, tienen un gen WT1 normal lo que no acarrea aumento del riesgo de tumor de Wilms.[32,91]
El tumor de Wilms en niños con síndrome WAGR se caracteriza por un exceso de enfermedad bilateral, restos nefrogénicos intralobulares, edad temprana en el momento del diagnóstico y un tipo histológico de predominio estromal en tumores con características HF.[16] Es posible que la discapacidad intelectual en el síndrome WAGR sea secundaria a la deleción de otros genes, como SLC1A2 o BDNF.[58]
- Síndrome de Denys-Drash. Esta afección se caracteriza por un síndrome nefrótico causado por esclerosis mesangial difusa, seudohermafroditismo XY y aumento del riesgo de tumor de Wilms (>90 %).
En este síndrome las variantes de WT1 por lo general son variantes de un aminoácido en los exones 8 y 9, que codifican la región de unión al DNA de WT1.[21]
- Síndrome de Frasier. Este síndrome se caracteriza por nefropatía progresiva causada por glomeruloesclerosis segmentaria focal, gonadoblastoma y seudohermafroditismo XY.
En este síndrome las variantes de WT1 suelen afectar el intrón 9 en el sitio KT, ello produce una variante de empalme alternativa, y por lo tanto, evitan la producción de la isoforma de WT1 +KTS que, por lo general, es más abundante.[23]
En los estudios de evaluación de las correlaciones genotípicas y fenotípicas de las variantes de WT1, se observó que el riesgo de tumor de Wilms es superior cuando hay variantes interruptoras (14 de 17 casos, 82 %), y el riesgo es inferior cuando hay variantes de aminoácido (27 de 67 casos, 42 %). El riesgo mas bajo se presenta cuando hay variantes en el sitio de empalme KTS (1 de 27 casos, 4 %).[88,89] El tumor de Wilms bilateral fue más común en los casos que tenían variantes interruptoras de WT1 (9 de 14 casos) en comparación con los casos que tenían variantes de cambio de sentido del gen WT1 (3 de 27 casos).[88,89] En estos estudios genómicos se corroboran las estimaciones previas de riesgo elevado de tumor de Wilms en los niños con síndrome de Denys-Drash, y de riesgo bajo de tumor de Wilms en niños con síndrome de Frasier.
GenCTNNB1
El gen CTNNB1 es uno de los genes alterados con mayor frecuencia en el tumor de Wilms; sus variantes se notifican en el 15 % de los pacientes con este tumor.[69,71,79,81,92] Las variantes de CTNNB1 producen la activación de la vía WNT, que cumple una función importante en el riñón en desarrollo.[93] Las variantes de CTNNB1 suelen acompañarse de variantes de WT1, y la mayoría de los casos de tumores de Wilms con variantes de WT1 exhiben al mismo tiempo una variante de CTNNB1.[79,81,92] La activación de la catenina β en presencia de una proteína WT1 intacta no es suficiente para promover la formación de un tumor porque las variantes de CTNNB1 casi nunca se encuentran en ausencia de una variante de WT1 o AMER1, excepto cuando se vinculan con una variante de MLLT1.[71,94] Las variantes de CTNNB1 son alteraciones tardías durante la formación del tumor de Wilms porque se encuentran en los tumores pero no en los restos nefrogénicos.[84]
GenAMER1(WTX) en el cromosoma X
El gen AMER1 está en el cromosoma X en Xq11.1 y se encuentra alterado en el 15 % al 20 % de los casos de tumor de Wilms.[70,71,79,95,96] Las variantes germinales de AMER1 causan una displasia ósea esclerosante ligada al cromosoma X, la osteopatía estriada congénita con esclerosis craneal (MIM300373).[97] Las personas con osteopatía estriada congénita no están predispuestas a presentar tumores, a pesar de tener variantes germinales de AMER1.[97] La proteína AMER1 participa en la degradación de la catenina β y en la distribución intracelular de la proteína APC.[94,98] Las alteraciones más comunes en el gen AMER1 son las deleciones que afectan una parte del gen AMER1 o el gen completo, y las menos comunes son las variantes de un solo nucleótido deletéreas.[70,79,95] La mayoría de los casos de tumor de Wilms con alteraciones en AMER1 presentan anormalidades epigenéticas en 11p15.[79]
Las alteraciones en AMER1 se distribuyen por igual entre hombres y mujeres, y la inactivación de AMER1 no tiene un efecto aparente en el cuadro clínico ni en el pronóstico.[70]
Regiones de control de impronta en el cromosoma 11p15 (WT2) y síndrome de Beckwith-Wiedemann
Otro locus del tumor de Wilms, el de WT2, está en la región de impronta del cromosoma 11p15.5. Cuando este exhibe una variante germinal, produce el síndrome de Beckwith-Wiedemann. Alrededor del 3 % de los niños con tumor de Wilms tiene cambios epigenéticos o genéticos germinales en el locus del regulador del crecimiento 11p15.5, sin ninguna otra manifestación clínica de sobrecrecimiento. Del mismo modo que los niños con síndrome de Beckwith-Wiedemann, en estos niños hay mayor incidencia de tumor de Wilms bilateral o tumor de Wilms familiar.[58]
Alrededor de un quinto de los pacientes con síndrome de Beckwith-Wiedemann que presentan un tumor de Wilms exhiben enfermedad bilateral, y además se ha observado enfermedad bilateral metacrónica.[32,33,34] En el National Wilms Tumor Study (NWTS) se notificó que la prevalencia del síndrome de Beckwith-Wiedemann es de casi el 1 % en los niños con tumor de Wilms.[4,34]
Cerca del 80 % de los pacientes con síndrome de Beckwith-Wiedemann tienen un defecto molecular en el dominio 11p15.[99] Se han identificado varios mecanismos moleculares subyacentes al síndrome de Beckwith-Wiedemann. Algunas de estas anomalías son genéticas (variantes germinales del alelo materno de CDKNIC, isodisomía uniparental paterna de 11p15, o duplicación de parte del dominio 11p15), pero es más común que sean epigenéticas (pérdida de metilación del ICR2 materno [genes CDKN1C y KCNQ1OT1] o ganancia de metilación del ICR1 materno [genes IGF2 y H19).[58,100]
Hay varios genes candidatos en el locus de WT2 que abarca dos dominios de impronta independientes: IGF2 con H19; y CDKN1C con KCNQ1OT1.[100] La LOH, que afecta de manera exclusiva el cromosoma materno, produce una activación regulada de los genes paternos activos y silenciamiento de los genes maternos activos. A menudo también se observa la pérdida o el cambio de la impronta para los genes (cambio en el estado de metilación) en esta región, lo que conlleva las mismas anomalías funcionales.[58,99,100]
Se demostró una relación entre el epigenotipo y el fenotipo en el síndrome de Beckwith-Wiedemann, y una diferencia en la tasa de cáncer en este síndrome de acuerdo con el tipo de alteración en la región 11p15.[101]
Hay cuatro subtipos moleculares principales de síndrome de Beckwith-Wiedemann que se caracterizan por correlaciones genotípicas y fenotípicas específicas:
- Ganancia de metilación en ICR1 (ICR1-GoM). La ICR1-GoM telomérica causa entre el 5 % y el 10 % de los casos. Esta alteración produce expresión bialélica del gen IGF2 (que por lo general solo se expresa a partir del alelo paterno) acompañada de disminución de la expresión del gen oncosupresor H19. La incidencia de tumor de Wilms es del 22,8 %.[102]
- Pérdida de metilación en ICR2 (ICR2-LoM). La ICR2-LoM causa el 50 % de los casos de síndrome de Beckwith-Wiedemann; esta alteración produce disminución en la expresión del gen CDKN1C que por lo general se expresa solo a partir del cromosoma materno. La incidencia del tumor es muy baja (2,5 %).[102]
- Disomía uniparental (DUP). Se observa una expresión alterada de ambos conjuntos de genes de impronta en la DUP mosaica del cromosoma 11p15.5, que explica del 20 % al 25 % de los casos. La incidencia de tumor de Wilms es del 6,2 %, seguida de hepatoblastoma (4,7 %) y carcinoma de glándula suprarrenal (1,5 %).[102] Menos del 1 % de los casos de síndrome de Beckwith-Wiedemann se producen a partir de reordenamientos cromosómicos que afectan la región 11p15.
- Variantes de CDKN1C. Las variantes de pérdida de función de CDKN1C heredadas por línea materna explican casi el 5 % de los casos. Este tipo se vincula con una incidencia de neuroblastoma de un 4,3 %.[102]
Se notificaron otros tumores como neuroblastoma o hepatoblastoma en pacientes con isodisomía paterna de 11p15.[26,30,103] En los pacientes con síndrome de Beckwith-Wiedemann, el riesgo relativo de hepatoblastoma es 2280 veces más alto que el de la población general.[34]
La pérdida de la impronta o la metilación génica es poco frecuente en otros locus, lo que respalda la especificidad de la pérdida de la impronta en 11p15.5.[104] El tumor de Wilms en niños de Japón y Asía del este, donde la incidencia es más baja que en niños blancos, no se relaciona con restos nefrogénicos ni con pérdida de impronta en IGF2.[105]
Otros genes y alteraciones cromosómicas
Otros genes y alteraciones cromosómicas que afectan la patogenia y las características biológicas del tumor de Wilms son los siguientes:
- 1q. La ganancia del cromosoma 1q se relaciona con un desenlace más precario y es el factor individual más poderoso para predecir el desenlace.[106,107] La ganancia del cromosoma 1q es una de las anomalías citogenéticas más comunes en el tumor de Wilms y se observa en alrededor del 30 % de los tumores.
En un análisis de tumores de Wilms con HF de 1114 pacientes participantes del NWTS-5 (COG-Q9401/NCT00002611) se encontró que el 28 % de los tumores exhibían ganancia de 1q.[106]
- La tasa de supervivencia sin complicaciones (SSC) a 8 años fue del 77 % en los pacientes con ganancia de 1q y del 90 % en aquellos sin ganancia de 1q (P < 0,001). En cada estadio de la enfermedad, la ganancia de 1q se relacionó con una SSC inferior.
- La tasa de supervivencia general (SG) a 8 años fue del 88 % en quienes tenían ganancia de 1q y del 96 % en aquellos sin ganancia de 1q (P < 0,001). La SG fue significativamente inferior en los casos con enfermedad en estadio I (P < 0,0015) y estadio IV (P = 0,011).
- También se notificaron resultados semejantes en el estudio WT 2001 de la International Society of Paediatric Oncology (SIOP) en 586 niños con tumor de Wilms.[107]
Un estudio incluyó una cohorte de tumor de Wilms de tipo HF con muchos pacientes en recaída. En este se encontró que la prevalencia de ganancia de 1q fue más alta en muestras de tumor de Wilms en recaída (75 %) que en las muestras primarias emparejadas (47 %).[108] La prevalencia en aumento de la ganancia de 1q en el momento de la recaída apoya su relación con un pronóstico precario y progresión de la enfermedad.
- 16q y 1p. Es posible que en los cromosomas 16q y 1p se encuentren otros genes supresores de tumores o genes de progresión tumoral, como lo indica la LOH de estas regiones en el 17 % y el 11 % de los casos de tumor de Wilms, respectivamente.[109]
- En estudios grandes del NWTS, los pacientes con pérdida en estos locus tumorales específicos presentaron tasas de supervivencia sin recaída y tasas de SG significativamente más precarias. En un estudio en curso del Children's Oncology Group (COG), se utiliza la combinación de la pérdida de 1p y 16q para seleccionar a los pacientes de tumor de Wilms y HF con el fin de administrarles un tratamiento más intensivo. Sin embargo, en un estudio del Reino Unido con más de 400 pacientes, no se encontró una relación significativa entre la deleción de 1p y un pronóstico adverso; pero se encontró un vínculo entre la LOH de 16q y un pronóstico adverso.[110]
- En un estudio italiano de 125 pacientes, se administró un tratamiento muy similar al del estudio del COG y se encontró un pronóstico significativamente más precario en los pacientes con deleciones de 1p, pero sin deleciones de 16q.[111]
Estos resultados contradictorios quizás se expliquen por la mayor repercusión pronóstica de la ganancia de 1q descrita antes. La LOH de 16q y 1p son menos relevantes como marcadores pronósticos independientes cuando hay una ganancia de 1q. Sin embargo, la ganancia de 1q y la LOH de 16q y 1p conservan su repercusión negativa sobre el pronóstico.[106] La LOH de 16q y 1p se originan por alteraciones cromosómicas complejas que conducen a LOH de 1q o ganancia de 1q. La alteración genética oncógena relevante es el cambio en 1q.[112]
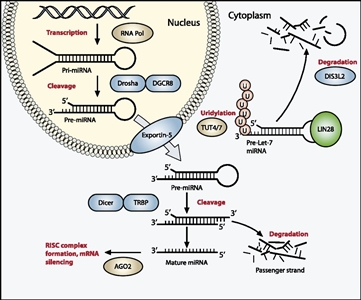
- miRNAPG. Se observaron variantes de determinados miRNAPG en alrededor del 20 % de los casos de tumor de Wilms, lo que al parecer perpetúa el estado del progenitor.[69,72,73,74,75] Los productos de estos genes dirigen la maduración de los miRNA desde los transcritos pre-miRNA hasta los miRNA funcionales citoplasmáticos (consultar la Figura 1).[113] El miRNAPG que está alterado con mayor frecuencia es DROSHA, que presenta una variante recurrente (E1147K) que afecta un residuo de enlace metálico en el dominio IIIb de la RNasa, y que explica cerca del 80 % de los tumores con alteraciones en DROSHA. Otros miRNAPG que se encuentran alterados en el tumor de Wilms son DGCR8, DICER1, TARBP2, DIS3L2 y XPO5. Por lo general, estas variantes son mutuamente excluyentes, deletéreas y alteran la expresión de los miRNA oncosupresores. Un sesgo sexual llamativo se señaló para las variantes de DGCR8 (ubicado en el cromosoma 22q11), pues 38 de 43 casos (88 %) surgieron en niñas.[72,73]
Se observaron variantes germinales de los miRNAPG de los genes DICER1 y DIS3L2; las variantes del primer gen causan el síndrome DICER1 y las variantes del segundo gen producen el síndrome de Perlman.
- El síndrome DICER1 a menudo surge debido a variantes interruptoras hereditarias del gen DICER1. Los tumores se forman después de que se adquiere una variante de cambio de sentido en un dominio del alelo remanente de DICER1 (dominio IIIb de la RNasa) responsable del procesamiento de los miRNA derivados de los brazos 5p de los pre-miRNA.[114] Los tumores relacionados con el síndrome DICER1 son el blastoma pleuropulmonar, el nefroma quístico, los tumores estromales de cordón sexual de ovario, el bocio multinodular y el rabdomiosarcoma embrionario.[114] El tumor de Wilms es una manifestación infrecuente del síndrome DICER1. En un estudio de 3 familias con síndrome DICER1 que incluían niños con tumor de Wilms, se encontró que 2 casos de tumor de Wilms exhibían la variante secundaria típica de DICER1 en el dominio IIIb de la RNasa.[115] En otro estudio, se encontraron variantes de DICER1 en 2 de 48 familias con tumor de Wilms familiar.[116] En extensos estudios de secuenciación de cohortes de casos de tumor de Wilms, también se observaron casos ocasionales con variantes de DICER1.[73,74]
- El síndrome de Perlman es un trastorno autosómico recesivo de sobrecrecimiento raro producido por variantes de DIS3L2, que codifica la ribonucleasa responsable de degradar el pre-let-7 miRNA.[38,117] Las inactivaciones germinales heterocigotas de DIS3L2 también se relacionan con la presentación de tumor de Wilms.[35] El pronóstico de los pacientes con síndrome de Perlman es precario, con una tasa de mortalidad neonatal alta. En una investigación de casos publicados de síndrome de Perlman (N = 28) en lactantes que sobrevivieron el periodo neonatal, se encontró que casi dos tercios presentaron tumor de Wilms y todos padecieron retraso del desarrollo. Las manifestaciones frecuentes son macrosomía fetal, ascitis y polihidramnios.[118]
- SIX1 y SIX2. Los genes SIX1 y SIX2 codifican factores de transcripción altamente homólogos que cumplen una función básica en el desarrollo renal temprano y que se expresan en el mesénquima metanéfrico en donde controlan la población mesenquimatosa progenitora. En los pacientes con tumor de Wilms, la frecuencia de las variantes de SIX1 es del 3 % al 4 %, y la frecuencia de las variantes de SIX2 es del 1 % al 3 %.[72,73]
- Prácticamente todas las variantes de SIX1 y SIX2 se ubican en el exón 1 y producen una variante caracterizada por un cambio de glutamina a arginina en la posición 177 (Q177R).
- Las variantes de los genes WT1, AMER1 y CTNNB1 son infrecuentes en los casos con variantes de los genes SIX1, SIX2 o de miRNAPG. Por el contrario, las variantes de SIX1 o SIX2 y las variantes de miRNAPG tienden a presentarse juntas.
- En muestras de tumor de Wilms de pacientes que no han recibido quimioterapia, las variantes de SIX1 y SIX2 se relacionan con el subtipo blastémico de riesgo alto y con la presencia de blastema indiferenciado.
- En un estudio de 82 casos de tumor de Wilms con HF, se identificaron variantes en el punto caliente Q177R de SIX1 con mayor frecuencia en las muestras tumorales de recaída (11 cases; 13,4 %) que en las muestras del momento del diagnóstico (4 %). En 45 casos que contaban con muestras del momento del diagnóstico y de la recaída, se encontraron 6 casos con SIX1Q177R en el momento de la recaída, 3 de los cuales no tenían SIX1Q177R en el momento del diagnóstico. Este resultado indica que esta variante no es necesaria para la formación del tumor en algunas personas con tumor de Wilms.[108]
- MLLT1. Alrededor del 4 % de los casos de tumor de Wilms tienen variantes en el dominio YEATS, altamente conservado, de MLLT1 (ENL), un gen involucrado en la elongación transcripcional producida por la RNA–polimerasa II durante el desarrollo temprano.[85] Las proteínas MLLT1 alteradas exhiben una alteración en la unión a los extremos de histonas acetiladas. Los pacientes con tumores que exhiben alteraciones de MLLT1 desde edades tempranas tienen una prevalencia alta de restos nefrogénicos intralobulares precursores, lo que sustenta un modelo en el que se presentan variantes activadoras de MLLT1 durante el desarrollo renal temprano que producen tumor de Wilms.
- TP53 (gen supresor de tumores). La mayoría de los casos de tumor de Wilms anaplásico exhiben variantes del gen supresor de tumores TP53.[119,120,121] El TP53 quizás sea útil como marcador de pronóstico desfavorable.[119,120]
En un estudio prospectivo de 118 pacientes con tumor de Wilms anaplásico difuso inscritos en el ensayo NWTS-5, se demostró que 57 pacientes (48 %) tenían variantes de TP53, 13 pacientes (11 %) tenían pérdida segmentaria del número de copias de TP53 sin variantes de este gen, y 48 pacientes (41 %) carecían de ambas anomalías (TP53 de tipo natural [wtTP53]). Todas las variantes de TP53 se detectaron mediante secuenciación sola. Los pacientes con enfermedad en estadio III o estadio IV y wtTP53 presentaron tasas de recaída y mortalidad significativamente más bajas que los pacientes con anomalías en TP53 (P = 0,00006 y P = 0,00007, respectivamente). El estado de TP53 no tuvo repercusión en los pacientes con tumores en estadio I o estadio II.[122]
- En un análisis detallado de un subconjunto de 39 pacientes con tumor de Wilms anaplásico difuso se observó que 7 pacientes (18 %) albergaban un wtTP53. En estos tumores con wtTP53 se demostró la expresión génica de la activación de la vía p53. En una revisión retrospectiva del resultado patológico de tumores con wtTP53 se observó un volumen muy bajo de anaplasia o ausencia de anaplasia en 6 de 7 tumores. Estos datos apoyan la función básica de la pérdida de TP53 en la formación de anaplasia en el tumor de Wilms, y respaldan la repercusión clínica significativa en pacientes con enfermedad anaplásica residual después de la cirugía.[122]
- FBXW7. Se identificó que el gen FBXW7, un componente de la ligasa de ubiquitina, es un gen supresor de tumores reconocido que presenta tasas bajas de alteraciones recurrentes en el tumor de Wilms y en otras neoplasias malignas. Las variantes de este gen se relacionaron con características histológicas tumorales de tipo epitelial.[123]; [124][Nivel de evidencia C1]
- TRIM28. El gen TRIM28 codifica una proteína con varios dominios que participa en la regulación de muchos procesos celulares y se considera un gen de predisposición al tumor de Wilms que se hereda de manera autosómica dominante. Las alteraciones en TRIM28 explican cerca del 8 % de los casos de tumor de Wilms familiar y 2 % de casos inespecíficos de tumor de Wilms.[54,125,126,127]; [124][Nivel de evidencia C1]
- Se ha observado una asociación fuerte entre las variantes de TRIM28 y el tumor de Wilms epitelial, la mayoría de las personas con una variante de TRIM28 presentan un tumor de Wilms con predominio de características histológicas epiteliales.[54,125,126]; [124][Nivel de evidencia C1]
- En una cohorte de 91 personas afectadas de 49 familias con árbol genealógico compatible con tumor de Wilms, se identificaron 33 personas que tenían variantes de predisposición al cáncer constitucionales, 21 de ellos presentaba una variante de TRIM28. Se observó un efecto fuerte del origen parental; los 10 casos evaluables presentaron variantes hereditarias transmitidas por vía materna.[124][Nivel de evidencia C1]
- La mayoría de los casos con variantes de TRIM28 exhibían variantes de cambio en el marco de lectura, terminadores o de corte y empalme en un alelo, en combinación con LOH en el otro alelo, lo que conlleva pérdida de la expresión de la proteína TRIM28 en el tumor. La tinción inmunohistoquímica para la pérdida de la expresión de la proteína TRIM28 se puede usar para identificar la mayoría de los pacientes que tienen variantes de TRIM28.[127]
- Síndrome de microdeleción de 9q22.3. Los pacientes con síndrome de microdeleción de 9q22.3 tienen riesgo elevado de presentar tumor de Wilms.[42] La región cromosómica con deleción germinal abarca el gen PTCH1, que está alterado en el síndrome de Gorlin (síndrome del carcinoma nevoide basocelular asociado con osteosarcoma). El síndrome de microdeleción de 9q22.3 se caracteriza por las manifestaciones clínicas del síndrome de Gorlin, así como por un retraso del desarrollo o discapacidad intelectual, craniosinostosis metópica, hidrocefalia obstructiva, macrosomía prenatal y posnatal, y convulsiones. Se informó sobre 5 pacientes que presentaban tumor de Wilms en el marco de una microdeleción constitucional de 9q22.3.[42,128,129]
- MYCN. Se han notificado alteraciones genómicas que afectan la red MYCN (por ejemplo, MYCN, MAX, MGA, NONO) en el 25 % al 30 % de los casos de tumor de Wilms.[108] Las alteraciones genómicas específicas asociadas con la red MYCN son las siguientes:
- La ganancia en el número de copias de MYCN se observó en cerca del 13 % de los casos de tumor de Wilms. La ganancia de MYCN fue más común en los casos anaplásicos (7 de 23 casos, 30 %) que en los casos no anaplásicos (11,2 %), y se relacionó con una supervivencia sin recaída (SSR) y supervivencia general más precarias, independientemente del tipo histológico.[130] La duplicación en tándem de MYCN se notificó en 11 de 82 (13 %) muestras de tumor de Wilms con HF.[108]
- La ganancia en el número de copias de MYCN de origen germinal se notificó en un caso de tumor de Wilms bilateral,[130] y la duplicación germinal de MYCN también se notificó en un niño con nefroblastomatosis bilateral prenatal y antecedentes familiares de nefroblastoma.[131]
- Se observaron variantes en el codón 44 (p.P44L) de MYCN en alrededor del 3 % al 4 % de los casos de tumor de Wilms en el momento del diagnóstico [130,132] y en el 8,5 % de los casos en el momento de la recaída.[108] En un estudio de 810 casos de tumor de Wilms, 24 (3 %) exhibía variantes en el punto caliente P44L de MYCN. La SSR fue significativamente inferior (68,6 %) en los pacientes con variantes de P44L que en los pacientes con MYCN natural (87,1 %).[132]
- La proteína de interacción con MYCN llamada MAX estaba alterada en el codón 60 (R60Q) en 7 de 782 casos de tumor de Wilms (0,9 %).[132] La SSR fue inferior en los pacientes con una variante en el punto caliente R60Q de MAX que en los pacientes con MAX natural.
- CTR9. En 4 de 36 árboles genealógicos de tumor de Wilms familiar se encontraron variantes inactivadoras germinales de CTR9.[57,133] El gen CTR9, ubicado en el cromosoma 11p15.3, es un componente clave del complejo del factor relacionado con la polimerasa de tipo 1 (PAF1c) que tiene múltiples funciones en la regulación de la RNA–polimerasa II y participa en la organogénesis embrionaria y la conservación de la pluripotencia de las células madre embrionarias.
- REST. Se encontraron variantes inactivadoras germinales de REST (codificador del factor de transcripción de silenciamiento RE1) en 4 árboles genealógicos de tumor de Wilms familiar.[77] El REST es un represor de la transcripción que afecta la diferenciación celular y el desarrollo embrionario. La mayoría de las variantes de REST se agrupan en la porción de REST que codifica el dominio de unión al DNA, y en el análisis funcional se observó que esas variantes afectan la represión transcripcional de REST. Cuando se hicieron pruebas para detectar variantes de REST, 9 de 519 personas con tumor de Wilms sin antecedentes familiares de la enfermedad obtuvieron un resultado positivo para una de estas variantes; algunos progenitores de estos pacientes también dieron positivo para una de estas variantes.[77] Estas observaciones permiten indicar que REST es un gen de predisposición al tumor de Wilms y que se asocia con casi un 2 % de estos tumores.
En la Figura 2 se resume el panorama genómico de una cohorte de pacientes con tumor de Wilms seleccionados debido a que presentaron recaída pese a exhibir HF.[85] Los 75 casos de tumor de Wilms con HF fueron agrupados en un análisis no supervisado de los datos de expresión génica; esto produjo 6 conglomerados. De los tumores para los que se contaba con datos de la expresión génica, 5 de 6 tumores tenían una alteración de MLLT1 y se ubicaron en el conglomerado 3, y 2 tumores presentaron simultáneamente variantes de CTNNB1. Este conglomerado también incluyó 4 tumores con una variante o deleción segmentaria pequeña de WT1, y todos tenían también una variante de CTNNB1 o una variante o deleción segmentaria pequeña de AMER1. Además abarcó un número considerable de tumores que conservaban la impronta de 11p15 (entre estos, todos los tumores con variantes de MLLT1). Los casos con alteraciones en miRNAPG estaban en el mismo conglomerado y eran mutuamente excluyentes de los casos con alteraciones de WT1, AMER1 o CTNNB1.

Alteraciones genómicas del tumor de Wilms en recaída
El tumor de Wilms en recaída conserva la mayoría de las alteraciones genómicas presentes en el momento del diagnóstico, aunque es posible que se presenten cambios en la prevalencia de alteraciones en genes específicos entre el momento del diagnóstico y de la recaída.[108] En un estudio del Children's Oncology Group se presentaron los datos de secuenciación del genoma completo (WGS) de muestras de tumores en recaída de 51 pacientes y muestras correspondientes del momento del diagnóstico para 45 de estos pacientes. Además se obtuvo un análisis de secuenciación dirigida para otros 31 pacientes que contaban con muestras del momento de la recaída. Los resultados principales fueron los siguientes:
- La prevalencia de ganancia de 1q en las muestras de tumores de Wilms en recaída (75 %) fue más alta que la observaba para los tumores en el momento del diagnóstico (47 %).[108] El aumento de la prevalencia de la ganancia de 1q en el momento de la recaída es compatible con su asociación con un pronóstico adverso y progresión de la enfermedad.
- Se identificaron variantes en el punto caliente SIX1Q177R con mayor frecuencia en las muestras tumorales de recaída (11 de 82 casos; 13,4 %) que en las muestras del momento del diagnóstico (4 %).[108] En los 45 casos que contaban con muestras del momento del diagnóstico y recaída, 6 casos presentaron la variante en el punto caliente SIX1Q177R en el momento de la recaída, 3 de los cuales no tenían la variante SIX1Q177R en el momento del diagnóstico. Esto es compatible con que la variante SIX1Q177R no se considere un evento de carcinogénesis temprana en algunos casos.[108]
- Se encontraron alteraciones genómicas en los genes asociados con la red MYCN el alrededor del 30 % de los casos de tumor de Wilms en recaída.[108] Las alteraciones más comunes en la red MYCN fueron la duplicación en tándem de MYCN (13 %) y las variantes en el punto caliente P44L de MYCN (11 %).
Los tumores de Wilms recidivantes o resistentes al tratamiento de 56 pacientes pediátricos se sometieron a secuenciación tumoral en el ensayo Pediatric MATCH del National Cancer Institute–Children's Oncology Group (NCI-COG). Durante el proceso se encontraron alteraciones genómicas que se consideraron de interés para el tratamiento en los grupos del estudio MATCH en 6 de 56 tumores (10,7 %). En 2 de 56 tumores (3,6 %), se encontraron variantes de BRCA2.[134]
Alteraciones genómicas en adultos con tumor de Wilms
El tumor de Wilms en pacientes mayores de 16 años es raro con una tasa de incidencia de menos de 0,2 casos por millón de personas por año.[2] Debido a esto hay pocos datos sobre las alteraciones genómicas que se observan en adultos con tumor de Wilms.
En un estudio de 14 pacientes con diagnóstico de tumor de Wilms y edad mayor a 16 años (intervalo, 17–46 años; mediana de edad, 31 años), se evaluaron las variantes exónicas de 1425 genes relacionados con el cáncer.[135]
- Se encontró que 5 pacientes (36 %) albergaban variantes BRAF V600E. Si bien las variantes BRAF V600E son muy infrecuentes en el tumor de Wilms en la niñez, se encuentran en el 90 % de los adenomas metanéfricos de riñón, una afección que suele ser benigna y que solo se presenta en adultos.[136]
- Los 5 adultos con tumor de Wilms y variante BRAF V600E exhibían áreas con mejor diferenciación idénticas a un adenoma metanéfrico situadas de manera adyacente a áreas compatibles en apariencia con el tumor de Wilms epitelial.
- Entre los 5 casos con variantes BRAF V600E, 2 presentaban variantes del promotor de TERT además de variantes de BRAF.
- Se observaron variantes de ASXL1 en 4 de 14 casos, que incluyeron 1 de 5 casos con variantes BRAF V600E y 3 de 9 casos sin variantes BRAF V600E. Las variantes de ASXL1 no son comunes en el tumor de Wilms infantil (alrededor del 2 % de los casos).[69]
- Entre los 9 tumores que no tenían variantes de BRAF, algunos presentaban alteraciones genómicas asociadas con el tumor de Wilms de la niñez (por ejemplo, ganancia de 1q y variantes de WT1 [n = 2]).
En otro informe se describieron tumores renales con superposición histológica de adenoma metanéfrico y tumor de Wilms epitelial.[137] Si bien la mayoría de los tumores de Wilms metanéfricos (5 de 9) que tenían áreas similares al adenoma metanéfrico no exhibían variantes BRAF V600E, se identificaron 4 casos con la variante BRAF V600E. De los casos con variantes BRAF V600E, 2 se presentaron en niños (3 y 6 años) y otros 2 en adultos.
Tumor de Wilms bilateral
Del 5 % al 10 % de las personas con tumor de Wilms presentan tumores bilaterales o multicéntricos. La prevalencia del compromiso bilateral es más alta en personas con síndromes de predisposición genética que en aquellos sin síndromes de predisposición. Por ejemplo, en 545 casos de tumor de Wilms bilateral, se encontraron variantes germinales patogénicas auténticas en el 22 % de los pacientes.[138] Las variantes de predisposición más comunes son las variantes de WT1 y la pérdida de la impronta de 11p15.[25,78]
La enfermedad bilateral puede ser sincrónica (ambos riñones afectados al mismo tiempo) o metacrónica (uno afectado después del otro) y se presenta en el 6,3 % y el 0,85 % de los pacientes con tumor de Wilms, respectivamente.[4] En general, los restos nefrogénicos perilobulares se relacionan con el tumor de Wilms bilateral sincrónico, mientras que los restos nefrogénicos intralobulares se relacionan más estrechamente con los tumores de Wilms metacrónicos.[139]
Los tumores de Wilms bilaterales con variantes de WT1 suelen presentarse de manera temprana en pacientes pediátricos (10 vs. 39 meses de vida en aquellos sin variante) y una frecuencia alta de variantes terminadoras en el exón 8 de WT1. El 3 % de los pacientes con tumor de Wilms bilateral tiene familiares afectados.[140]
El análisis genómico de tejido renal de tumor de Wilms bilateral indica que antes de la divergencia de los primordios renales izquierdo y derecho se produjo una expansión clonal al comienzo de la nefrogénesis de lesiones precursoras de aspecto normal pero con anormalidades genéticas.[68]
Exámenes de detección para niños con predisposición al tumor de Wilms
El objetivo principal de los exámenes de detección es permitir la identificación temprana de un tumor pequeño y localizado (estadio I o II), mejorar el pronóstico y utilizar un tratamiento menos intensivo (como para facilitar la cirugía conservadora de nefronas).[141] Los niños con un aumento significativo de la predisposición a presentar un tumor de Wilms (por ejemplo, la mayoría de los niños con síndrome de Beckwith-Wiedemann u otros síndromes de sobrecrecimiento, síndrome WAGR, síndrome de Denys-Drash, aniridia esporádica o hemihipertrofia aislada) se suelen examinar con ecografía cada 3 meses hasta que alcanzan por lo menos los 8 años de edad.[91,141]
Se han recomendado programas de exámenes de detección tumoral para cada síndrome de sobrecrecimiento. Estos programas se basaron en las publicaciones que indican la edad y la incidencia del tipo de tumor, así como en las recomendaciones del American Association for Cancer Research (AACR) Childhood Cancer Predisposition Workshop de 2016. Aunque están apareciendo datos sobre diferentes riesgos de cáncer según subgrupos genéticos o epigenéticos para ciertos síndromes, y aunque se formularon recomendaciones específicas para los subgrupos en Europa, estas prácticas no se han adoptado en los Estados Unidos. El comité del grupo de trabajo de AACR propuso un abordaje de detección uniforme para todos los síndromes relacionados con un riesgo de tumor de Wilms superior al 1 %. También se recomiendan otros exámenes de detección para el hepatoblastoma mediante la medición de la alfafetoproteína (AFP) sérica y la ecografía para los pacientes con síndrome de Beckwith-Wiedemann, trisomía 18 y síndrome de Simpson-Golabi-Behmel.[142]
A partir de una búsqueda en la bibliografía de pacientes en el espectro de Beckwith-Wiedemann y tumor de Wilms donde la edad en el momento del diagnóstico se comparó con datos recabados mediante el programa de Surveillance, Epidemiology, and End Results (SEER), se encontró que los exámenes de detección de pacientes en el espectro de Beckwith-Wiedemann reducen de manera significativa la edad y el estadio en el momento del diagnóstico en esta población. Hacer exámenes de detección hasta los 7 años de edad es eficaz para detectar cerca del 95 % de todos los tumores de Wilms en niños en el espectro de Beckwith-Wiedemann. Los exámenes de detección hasta los 30 meses de vida también podrían ser útiles para pacientes con ICR2-LoM, lo que es congruente con las recomendaciones sobre la detección del hepatoblastoma en esta población.[143]
- Síndrome de Beckwith-Wiedemann. Alrededor del 8 % de los pacientes con síndrome de Beckwith-Wiedemann presentará una neoplasia maligna, más a menudo un tumor de Wilms o un hepatoblastoma, aunque también se presentan tumores de glándula suprarrenal.[102]
La detección de hepatoblastoma o tumores de glándula suprarrenal mediante ecografía abdominal y AFP sérica suele comenzar desde el nacimiento o cuando se diagnostica el síndrome y continúa hasta los 4 años de edad Después de los 4 años, se habrán presentado la mayoría de los hepatoblastomas y las pruebas con imágenes se pueden limitar a una ecografía renal, que es más rápida y no exige ayuno previo.[144]
El uso de exámenes de detección para el tumor de Wilms suele continuar hasta los 8 años de edad. Se recomienda que un especialista (genetista u oncólogo pediátrico) realice un examen físico dos veces por año, y se debe incluir educación continua sobre las manifestaciones tumorales para reforzar la justificación del uso de los exámenes de detección y el cumplimiento con el régimen de detección.[142]
Se dispone de directrices sobre los exámenes de detección del tumor de Wilms en pacientes con síndrome de Beckwith-Wiedemann sometidos a tipificación de subtipos moleculares[102] Los cuatro subtipos moleculares principales del síndrome de Beckwith-Wiedemann (ICR1-GoM, ICR2-LoM, DUP y con variante de CDKN1C) se caracterizan por correlaciones específicas entre el genotipo y el fenotipo, incluso el riesgo tumoral. Para obtener más información sobre los subtipos moleculares, consultar la sección Características genómicas del tumor de Wilms.
A continuación se describen los exámenes de detección propuestos para los subtipos moleculares específicos del síndrome de Beckwith-Wiedemann:
- Los pacientes con un defecto de la región ICR1 (ICR1-GoM) y DUP deben someterse a ecografía abdominal cada 3 meses hasta los 8 a 10 años. El examen clínico del abdomen y la masa muscular se realiza mensualmente durante el primer año y luego a intervalos de 3 meses, entre las ecografías, hasta los 6 años de edad.
- En los pacientes con pérdida de impronta en ICR2 (ICR2-LOM), se realiza una ecografía abdominal en el momento del diagnóstico clínico o molecular. Solo los pacientes con organomegalia o hemihipertrofia grave requieren vigilancia mediante ecografías. Se realiza una examen físico mensual durante los primeros 2 años, seguido de un examen físico cada 3 a 6 meses hasta los 6 años.
- Los pacientes con una variante de CDKN1C no tienen un aumento del riesgo de tumor de Wilms. No hay datos que justifiquen el uso de exámenes de detección de rutina.
- Síndrome WAGR (o espectro WAGR). Los pacientes con espectro WAGR tienden a presentar tumor de Wilms a una edad más temprana que los pacientes con tumor de Wilms no sindrómico. Con los datos del WAGR Syndrome Patient Registry (n = 91), se calculó que la mediana de edad notificada en el momento de la presentación inicial del tumor de Wilms o de restos nefrogénicos aislados fue de 19 meses (intervalo, 11–28 meses). Todos los pacientes con tumor de Wilms notificado presentaron el primer tumor de Wilms antes de los 8 años, y el 95 % de los pacientes presentaron el tumor antes de los 5 años.[13] Otros investigadores notificaron que cerca del 20 % de los pacientes presentaron el primer tumor de Wilms después de los 4 años.[15,16]
Varios pacientes recibieron el diagnóstico de tumor de Wilms después de los 7 a 8 años de edad o presentaron una recaída años después del diagnóstico inicial. Algunos casos considerados como recaídas en realidad se trataban de una enfermedad de novo en el riñón contralateral.[16] En el WAGR Syndrome Patient Registry, un participante presentó una recaída tardía a los 19 años y 7 meses de edad; esto ocurrió más de 17 años después del primer diagnóstico de tumor de Wilms y representó la tercera aparición.[13]
Los investigadores del SIOP informaron sobre el beneficio de la vigilancia en una cohorte de 43 pacientes con síndrome WAGR y tumor de Wilms o nefroblastomatosis inscritos en estudios de tratamiento del SIOP. De 39 pacientes, 27 (69 %) fueron asintomáticos y los tumores se detectaron mediante vigilancia, mientras que 12 pacientes (31 %) presentaron una masa abdominal palpable o visible, u otros síntomas. De estos 12 pacientes, 2 no habían sido diagnosticados con síndrome WAGR. Los tumores detectados mediante vigilancia tenían un volumen significativamente más reducido en comparación con los tumores de pacientes sintomáticos (18 vs. 375 ml; P = 0,001), lo que permitió una tasa alta de cirugía con conservadora de nefronas (85 %).[145] Los autores recomiendan el uso de quimioterapia preoperatoria como tratamiento para los pacientes con síndrome WAGR con el fin de facilitar la cirugía conservadora de nefronas. Esta cirugía puede mejorar los desenlaces de los pacientes con enfermedad renal crónica relacionada con el síndrome WAGR.[146] Se notificó que la quimioterapia preoperatoria disminuye el tamaño del tumor en el 50 % de los pacientes con WAGR.[145]
Las opciones de vigilancia en la población con síndrome WAGR a partir de los 8 años de edad se deben analizar con la familia del paciente y el equipo multidisciplinario de atención de la salud para determinar el programa de seguimiento adecuado para la vigilancia del tumor de Wilms. Se deben considerar factores como los antecedentes del paciente y la presencia de restos nefrogénicos y nefroblastomatosis.[13]
- Hemihipertrofia (también conocida como sobrecrecimiento lateralizado o hemihiperplasia). Los niños con hemihipertrofia aislada también tienen riesgo de presentar tumores de hígado, tumores de glándula suprarrenal y tumor de Wilms (riesgo, 3–4 %). Se indica el uso de exámenes de detección con ecografía abdominal y AFP sérica hasta los 4 años de edad. Después de los 4 años, se habrán presentado la mayoría de los hepatoblastomas y las pruebas con imágenes se pueden limitar a una ecografía renal, que es más rápida y no exige ayuno previo.[142]
La hemihipertrofia se puede presentar como parte de un síndrome (con mayor frecuencia, el síndrome de Beckwith-Wiedemann) o un fenómeno aislado. El Beckwith-Wiedemann Syndrome International Consensus Group indicó que las personas con fenotipo florido del síndrome de Beckwith-Wiedemann y aquellas con hemihipertrofia aislada que tienen hallazgos moleculares similares a los del síndrome de Beckwith-Wiedemann deben considerarse parte del espectro de este síndrome y deben recibir el tratamiento acuerdo con el subtipo que corresponda. Se pueden considerar pruebas moleculares para pacientes con hemihipertrofia aislada según el sistema de puntuación clínica propuesto por el Beckwith-Wiedemann Syndrome International Consensus Group.[147]
Es posible que los niños con hemihipertrofia aislada y pruebas moleculares negativas no necesiten vigilancia porque quizás el riesgo es muy bajo. Sin embargo, se necesitan más estudios de cohortes grandes de niños sometidos a pruebas moleculares con hemihipertrofia aislada para determinar el riesgo.[91,147]
- Aniridia esporádica. Los recién nacidos con aniridia esporádica deben someterse a pruebas moleculares para el análisis de deleción de PAX6 y WT1, que son compatibles con el síndrome WAGR. Cerca del 30 % de los pacientes con aniridia esporádica tienen WAGR.[148] Si se observa una deleción en WT1, el niño se deberá hacer un examen de detección con ecografía cada 3 meses hasta los 8 años, y se debe informar a los padres sobre la necesidad de identificar y tratar de manera oportuna un tumor de Wilms.[91,149,150]
- Descendientes de sobrevivientes de tumor de tumor Wilms bilateral. Aunque no se conoce el riesgo de tumor de Wilms en los hijos de ambos sexos de personas sobrevivientes de tumor de Wilms bilateral y, es probable que el riesgo varíe según el gen en el que se produjo la variante, algunos expertos recomiendan que estos niños y niñas se sometan a exámenes de detección con ecografías en serie cada 3 meses hasta cumplir los 8 años de edad.[76]
- Síndrome de Bohring-Opitz. El síndrome de Bohring-Opitz es una afección genética rara relacionada con variantes de ASXL1. En general, cerca del 7 % de las personas con síndrome de Bohring-Opitz presentan tumores de Wilms.[151] Por este motivo, se indica el uso de exámenes de detección con ecografía abdominal cada 3 a 4 meses durante los primeros 8 años de vida.[47]
- Síndrome de Simpson-Golabi-Behmel. Los varones afectados por el síndrome de Simpson-Golabi-Behmel que tienen variantes o deleciones de GPC3 presentan un riesgo cercano al 10 % de tumor Wilms. En los varones con síndrome de Simpson-Golabi-Behmel se recomienda el uso rutinario de exámenes de detección tumoral acordes con la edad, que incluyan ecografía abdominal, uroanálisis y marcadores bioquímicos, aunque no se ha determinado su beneficio real. Antes se pensaba que las mujeres portadoras no tenían un riesgo elevado de tumor de Wilms y no necesitaban vigilancia. Sin embargo, hay informes de casos poco frecuentes en los que el síndrome de Simpson-Golabi-Behmel tuvo una expresión clínica significativa en mujeres y se presentaron casos de tumores de Wilms.[91] En las mujeres con afectación indudable, se deben considerar los exámenes de detección para los tumores embrionarios, incluso los tumores de Wilms.[152]
- Síndrome de Klippel-Trénaunay. El riesgo de tumor de Wilms en los niños con síndrome de Klippel-Trénaunay (síndrome de sobrecrecimiento unilateral de una extremidad) no fue diferente al riesgo en la población general cuando se evaluó en la base de datos del National Wilms Tumor Study (NWTS). No se recomienda la vigilancia ecográfica de rutina.[153]
- Síndrome de Perlman. El síndrome de Perlman es un síndrome de sobrecrecimiento congénito raro de herencia autosómica recesiva. Es posible establecer el diagnóstico molecular al detectar la presencia de variantes inactivadoras del gen DIS3L2 del cromosoma 2q37.1. Morirán el 53 % de los niños en el período neonatal. Los riñones exhiben nefroblastomatosis en cerca del 75 % de los casos. La incidencia del tumor de Wilms es del 64 % en lactantes que sobreviven más allá del período neonatal.[38] Se recomienda que estos pacientes reciban vigilancia regular similar a la que se ofrece a los pacientes con síndrome de Beckwith-Wiedemann.[142]
- Síndrome DICER1. El nefroma quístico se observa en el 10 % de las familias que tienen blastoma pleuropulmonar, por lo general antes de los 4 años. Es posible que haya progresión a un sarcoma anaplásico de riñón, pero esto es infrecuente. El síndrome DICER1 acarrea un riesgo elevado de tumor de Wilms, que no es una consecuencia de un nefroma quístico anterior. La vigilancia consiste en una ecografía abdominal, que comienza con una tomografía computarizada (TC) torácica inicial para detectar el blastoma pleuropulmonar, y se realiza cada 6 a 12 meses hasta los 8 años de edad. Es posible que se continúe con la vigilancia una vez al año hasta los 12 años de edad, según el paciente.[154] Trece años es la mayor edad notificada de diagnóstico de tumor de Wilms en un portador de variante de DICER1.[116,155] El objetivo de la vigilancia es identificar los nefromas quísticos cuando son pequeños y todavía es posible realizar una cirugía conservadora de nefronas, ya que los tumores que progresan a sarcoma anaplásico de riñón tienen tasas de morbilidad más elevadas. Dado que el sarcoma anaplásico de riñón se diagnostica en intervalos de edad más amplios que el nefroma quístico, debe considerarse la posibilidad de ampliar la detección mediante ecografía abdominal hasta los 12 años. Este intervalo de tiempo es el período de mayor riesgo para el sarcoma anaplásico de riñón (90 % de los casos de línea germinal presumiblemente detectados).[156]
- Variante germinal patogénica o probablemente patogénica en los genes de predisposición al tumor de Wilms (CTR9, REST, TP53, TRIM28) en ausencia de características del síndrome o una historia familiar que indique algún síndrome específico de predisposición al cáncer. Se debe considerar la vigilancia durante todo el período de riesgo alto de tumor de Wilms (por lo general hasta los 8 años de edad), pero puede variar según la afección.[157]
Asesoramiento genético
El tumor de Wilms se presenta asociado a una predisposición de la línea germinal subyacente en el 10 % al 15 % de los casos. Se recomienda una remisión a consulta de genética a todos los niños con tumor de Wilms que tengan antecedentes familiares de cáncer, compromiso renal bilateral o presencia de características específicas del síndrome.[157]
El estudio McGill Interactive Pediatric OncoGenetic Guidelines (MIPOGG) tiene como objetivo desarrollar una herramienta de eHealth para ayudar a los médicos a identificar a los niños con mayor riesgo de tener un síndrome de predisposición al cáncer. A partir de una revisión exhaustiva de la literatura, se desarrolló un algoritmo de decisión específico para el tumor de Wilms. Este algoritmo se compone de 5 criterios específicos del tumor (edad <2 años, bilateralidad o multifocalidad, tipo histológico de predominio estromal, restos nefrogénicos y características de sobrecrecimiento) y criterios universales, que incluyen características de una historia familiar compatible con un síndrome de predisposición al cáncer y anomalías congénitas. Esta herramienta se aplicó de forma retrospectiva a 180 pacientes pediátricos consecutivos con tumor de Wilms, diagnosticados o tratados en el Hospital for Sick Children (1997–2016) que se sometieron a pruebas de diagnóstico molecular dirigido.[158]
- La aplicación del algoritmo generó una recomendación binaria a favor o en contra de la derivación a consulta genética para las evaluaciones del síndrome de predisposición al cáncer. El algoritmo identificó al 100 % de los niños con tumor de Wilms y un síndrome de predisposición al cáncer confirmado (n = 27).
- La edad menor de 2 años, la bilateralidad o multifocalidad y las anomalías congénitas se relacionaron de manera importante con variantes patogénicas de WT1.
- La presencia de más de una característica de sobrecrecimiento se relacionó directamente con el síndrome de Beckwith-Wiedemann.
- El tipo histológico de predominio estromal no contribuyó al reconocimiento del síndrome de predisposición al cáncer (y se eliminó de los criterios esenciales). En combinación con otras características compatibles, las características histológicas estromales pueden aumentar la probabilidad de identificar un trastorno relacionado con WT1.
- Se están planeando pruebas de esta herramienta de forma prospectiva en pacientes con diagnóstico de tumor de Wilms que se someten a una secuenciación genómica completa del DNA germinal.
Si se descubre que un niño alberga una variante patogénica o probablemente patogénica en un gen de predisposición al tumor de Wilms, también se le puede ofrecer pruebas a sus padres y familiares cercanos. Se debe aconsejar a las personas afectadas sobre el riesgo de neoplasias y manifestaciones oncológicas adicionales, según corresponda, así como sobre el riesgo para la futura descendencia.[157]
Características clínicas del tumor de Wilms
El cuadro clínico inicial de la mayoría de los pacientes con tumor de Wilms incluye una masa abdominal asintomática percibida por un progenitor o por el pediatra durante una consulta del niño sano. A veces se encuentran tumores renales durante los exámenes de detección periódicos en niños con síndromes de predisposición reconocidos. Las manifestaciones clínicas incluyen las siguientes:
- Masa, distensión o dolor abdominal. La mayoría de los niños presentan una masa grande en el flanco que se advierte cuando se bañan o se visten. Cuando se detecta en el examen físico, se puede observar que la masa no se mueve con la respiración a diferencia de la esplenomegalia. Dolor abdominal, en el 40 % de los niños.
- Sangre en la orina. Cerca del 18 % de los niños con tumor de Wilms presenta hematuria macroscópica en el cuadro clínico inicial, y el 24 % de los pacientes exhibe hematuria microscópica.[159]
- Hipertensión. Alrededor del 25 % de los niños tiene hipertensión en el cuadro clínico inicial, que se debe a la activación del sistema de la renina y la angiotensina.
- Hipercalcemia. Algunas veces, se observa hipercalcemia sintomática en el momento de la presentación de tumores rabdoides.
- El 10 % de los casos tiene síntomas constitucionales como fiebre, anorexia y pérdida de peso.
Es posible que los niños con tumores de Wilms u otras neoplasias malignas de riñón acudan a una consulta médica a raíz de las siguientes manifestaciones:
- Obstrucción vascular o metástasis, incluso síntomas pulmonares como disnea causados por metástasis pulmonar.
- Dolor abdominal causado por una metástasis hepática, vasos prominentes en la pared abdominal o varicocele producto de una obstrucción en la vena cava inferior.
- Embolia pulmonar (poco frecuente).
- Es posible que los pocos niños que presentan hemorragia subcapsular presenten distensión abdominal rápida, anemia y dolor intenso.
Evaluación diagnóstica y estadificación del tumor de Wilms
El Children's Oncology Group Diagnostic Imaging Committee y el Society for Pediatric Radiology Oncology Committee publicaron un informe con recomendaciones para las pruebas con imágenes de tumores renales infantiles.[160] Las pruebas y los procedimientos que se utilizan para diagnosticar y estadificar el tumor de Wilms y otros tumores renales infantiles son las siguientes:
- Examen físico y antecedentes. Los niños con una masa renal se evalúan cuidadosamente para detectar signos de síndromes relacionados, como aniridia, retraso del desarrollo, hipospadias, criptorquidia, pseudohermafroditismo, sobrecrecimiento e hemihipertrofia.
- Recuentos sanguíneos completos.
- Pruebas del funcionamiento hepático.
- Pruebas del funcionamiento renal.
- Análisis de orina.
- Imágenes abdominales.
- Radiografía abdominal.
- Ecografía abdominal. La ecografía abdominal se suele realizar antes de una exploración más definitiva con TC o imágenes por resonancia magnética (IRM) con contraste del abdomen. Este procedimiento es innecesario después de que se realice el estudio diagnóstico definitivo.[160]
- TC con contraste (no se necesita contraste oral) o IRM de abdomen y pelvis (con contraste intravenoso o sin este).[160]
- La TC abdominal corrobora el origen renal de la masa y permite determinar si hay tumores bilaterales.[161] Alrededor del 5 % de las masas renales que se cree que son tumores de Wilms según los hallazgos clínicos y radiológicos finalmente se diagnostican como otra afección.[162]
- En una revisión de niños con tumor de Wilms bilateral se demostró que la TC helicoidal pasó por alto solo el 0,25 % de los tumores bilaterales, todos ellos eran tumores pequeños.[163]
- La evaluación preoperatoria con imágenes del compromiso intravascular del tumor de Wilms es fundamental para guiar el tratamiento. Entre los pacientes con tumor de Wilms, el 4 % presenta compromiso de la vena cava inferior o de las aurículas, y el 11 % presenta compromiso de la vena renal, lo que quizás afecte el tratamiento. La embolización de un trombo desde la vena cava hasta la arteria pulmonar es poco frecuente, pero puede causar la muerte; los trombos se deben identificar antes de la cirugía con el fin de prevenir la embolización y guiar el tratamiento. En un informe del COG se demostró que una TC permite identificar con exactitud los trombos cavo-auriculares, lo que evita la necesidad de una ecografía si ya se realizó una TC.[164]
- La ascitis en el fondo de saco es el mejor predictor de la rotura preoperatoria del tumor de Wilms, independientemente de la atenuación. Cuando hay ascitis, la presencia de trabeculación de grasa alrededor del tumor y de líquido retroperitoneal predice en gran medida una rotura.[161]
- La preocupación por la TC es la exposición a la radiación, pero el procedimiento es rápido, permite imágenes continuas del tórax y el abdomen, tiene una especificidad moderada para la detección del derrame preoperatorio, puede ayudar a distinguir los restos nefrogénicos del tumor de Wilms y proporciona excelentes detalles pulmonares.[161,165]
- El principal inconveniente de la IRM abdominal es que a menudo se necesita sedación de moderada a profunda en los niños pequeños. Sin embargo, proporciona un excelente detalle de los órganos en pacientes con compromiso bilateral o metástasis hepáticas. Se prefiere la IRM abdominal para una mejor evaluación de los posibles restos nefrogénicos y su distinción del tumor de Wilms verdadero.[166] Si la decisión es realizar una IRM abdominal, primero se debe realizar una TC de los pulmones para evitar el oscurecimiento de las bases pulmonares por atelectasia.[167] La IRM es la modalidad por imágenes preferida en niños con tumores de Wilms bilaterales conocidos o predisposición tumoral bilateral conocida.[167]
- TC del tórax. Alrededor del 15 % al 20 % de los pacientes presentarán metástasis. Los sitios comunes de metástasis para el tumor de Wilms son los pulmones (85 %), el hígado (10 %), así como los huesos y la columna vertebral (infrecuente). La TC es el método más sensible para la detección de nódulos pulmonares metastásicos. En las TC de tórax iniciales, se prefiere el uso de contraste intravenoso yodado porque permite la evaluación simultánea del parénquima pulmonar, el sistema vascular regional y otras estructuras del mediastino.[160] Aproximadamente el 7,5 % de los pacientes presentan derrame pleural.[168][Nivel de evidencia C1]
- No es necesario obtener una radiografía del tórax si ya se hizo una TC torácica al inicio.
- Tomografía por emisión de positrones con flúor F 18-fludesoxiglucosa (TEP con 18F-FDG). La TEP no se usa de forma rutinaria en el tumor de Wilms, aunque el tumor de Wilms presenta afinidad por la 18F-FDG. Las imágenes de TEP-TC con 18F-FDG añaden información con utilidad clínica a las que se obtienen por las imágenes de TC convencionales. Esto a veces es muy útil para los pacientes con enfermedad bilateral o que reciben quimioterapia preoperatoria. La TEP-TC con 18F-FDG destaca las áreas ávidas de FDG en el tumor y las metástasis, que se corresponden con la enfermedad activa confirmada histológicamente.[169]
- La gammagrafía ósea o las imágenes transversales de otros sitios se reservan para los pacientes con signos o síntomas de metástasis extrapulmonares a distancia.
- Evaluación diagnóstica de la enfermedad de Von Willebrand. Alrededor del 1 % al 4 % de los pacientes con tumor de Wilms adquieren una forma de enfermedad de Von Willebrand, aunque muchos son asintomáticos; los multímeros de Von Willebrand se unen al tumor de Wilms lo que reduce la concentración plasmática de multímeros hasta cantidades bajas.[170] Algunos médicos recomiendan la evaluación de la enfermedad de Von Willebrand antes de la cirugía, porque, aunque es poco común, a veces acarrea riesgos importantes de hemorragia y se puede tratar de manera preventiva. La enfermedad de Von Willebrand adquirida en el contexto del tumor de Wilms por lo general se resuelve una vez que se inicia la quimioterapia o se reseca el tumor.[171]
- Biopsia o resección. En los niños con una masa renal que clínicamente parece ser un tumor de Wilms en estadio I o estadio II, no se realiza una biopsia para evitar que las células tumorales se diseminen durante el procedimiento de la biopsia. En estos casos la biopsia llevaría a reclasificar el paciente en un estadio III. En lugar de biopsia se hace una nefrectomía (en América del Norte) o se administra quimioterapia (en Europa). Por tal motivo, el diagnóstico patológico recién se determina cuando se examina la pieza de la nefrectomía.
A veces, se indica una biopsia de una masa renal si el aspecto radiográfico de la masa es atípico para un tumor de Wilms, y el paciente no se va a someter de inmediato a una nefrectomía. Ante la imposibilidad de realizar una nefrectomía primaria, se necesita una biopsia, ya sea abierta o por múltiples punciones con aguja gruesa. Las contraindicaciones para la nefrectomía primaria son las siguientes:
- Extensión de un trombo tumoral hasta el nivel de las venas hepáticas. En estos pacientes se debe considerar la resección tumoral después de quimioterapia neoadyuvante cuando hay indicios de regresión del trombo de la vena cava independientemente del grado de respuesta del tumor primario.
- Tumor que compromete estructuras contiguas de manera que la única forma de extirpar el tumor renal es extirpando la otra estructura comprometida (por ejemplo, el bazo, el páncreas y el colon, excepto la glándula suprarrenal o el diafragma). Si bien los tumores de Wilms suelen adherirse a los órganos adyacentes, en la mayoría de los casos, no hay una invasión tumoral franca y es posible la disección del tumor con liberación de estos órganos. Por lo general, no se justifica la resección en bloque (por ejemplo, hepatectomía parcial). Si la extirpación de una sección pequeña del diafragma, el músculo psoas, o un extremo del páncreas permite la extirpación del tumor intacto, esta opción se considera inocua y apropiada.
- Si al criterio del cirujano se considera que la nefrectomía conllevaría morbilidad o mortalidad significativas o innecesarias, derrame tumoral significativo o tumor residual.[172]
- Si hay compromiso pulmonar debido a metástasis pulmonares extensas o, en casos infrecuentes, enfermedad hepática.
Si el primer procedimiento de un niño es una biopsia, entonces se clasifica en estadio III porque tiene tumores residuales macroscópicos.
Es posible usar el tejido de la biopsia de un tumor de Wilms inoperable obtenido antes de la quimioterapia para el análisis histológico y con el fin de tomar decisiones sobre el tratamiento inicial. Sin embargo, el empleo de la biopsia para determinar el tipo histológico de un tumor inoperable es polémico porque la biopsia a veces produce diseminación local del tumor y no permite determinar la clasificación histológica del tumor de Wilms.[172]
La heterogeneidad tumoral dificulta la detección del tipo histológico anaplásico en cualquier muestra de biopsia. Los datos de los estudios NWTS-4 y NWTS-5 (COG-Q9401/NCT00002611) demostraron que, debido a la heterogeneidad histológica del tumor de Wilms, un número considerable de pacientes exhibe un tipo histológico anaplásico que se pasa por alto durante la biopsia inicial, independientemente de si es una biopsia por punción con aguja gruesa o por incisión,[173] aunque la anaplasia se identificó en el momento de la cirugía definitiva después de la quimioterapia.
En ocasiones, se cambia el estadio y el tratamiento inicial cuando se detecta una lesión renal contralateral en un niño con tumor de Wilms; ello denota una función de un abordaje de conservación renal sin cirugía inicial. La detección de lesiones renales contralaterales es importante en las imágenes iniciales porque ya no se recomienda la exploración intraoperatoria sistemática del riñón contralateral según los resultados del estudio NWTS-4.[163,174] Se debe considerar el tratamiento como si fuese un tumor de Wilms bilateral cuando en las pruebas con imágenes iniciales se observa un proceso bilateral. Si el origen de la otra lesión es indeterminado, se debe considerar una evaluación patológica de esa lesión antes de proceder con una nefrectomía.[163,174]
Los niños que tienen tumor de Wilms bilateral suelen tratarse sin una biopsia.[175] La biopsia se puede evitar si el niño tiene la edad típica y el tumor tiene la apariencia radiográfica habitual. Esto se evaluó en el estudio del COG AREN0534 (NCT00945009) en el que 187 de 189 pacientes con tumor de Wilms se trataron de manera inicial sin biopsia. Si al cabo de 6 semanas de tratamiento, la respuesta era inferior al 30 % conforme a los criterios RECIST1.1, entonces se obtenían biopsias bilaterales para determinar la presencia de anaplasia, diferenciación estromal y cambios rabdomiomatosos. Si se detectaba anaplasia, se modificaba el tratamiento con quimioterapia. Si se detectaba diferenciación estromal o cambios rabdomiomatosos, era improbable que el uso de quimioterapia adicional llevase a la reducción de la masa tumoral y se recomendó un abordaje quirúrgico definitivo.[175]
Cuando se sospecha la presencia de tumor de Wilms, se necesitan otros estudios de estadificación preoperatoria con el fin de evaluar el estado ganglionar, la extensión intravascular y la rotura del tumor de Wilms.[162]
- Se exige el muestreo de ganglios linfáticos para la estadificación local de todos los pacientes con tumor de Wilms. Se ha demostrado que los ganglios linfáticos tienen valor pronóstico importante para la supervivencia a corto y largo plazo. La inspección macroscópica es muy inexacta, con una tasa de negativos falsos del 31,3 % y una tasa de positivos falsos del 18,1 %.[176]
- Extensión intravascular del tumor de Wilms. La evaluación preoperatoria de la extensión intravascular del tumor de Wilms es fundamental para orientar el tratamiento. La presencia de trombos tumorales intravenosos en el lumen de la vena renal, la vena cava inferior y la aurícula derecha se notificó hasta en un 11,3 % de los pacientes con tumor de Wilms, y puede conducir a diferencias de tratamiento.
En América del Norte, la estadificación local del tumor de Wilms se determina mediante TC o IRM del abdomen y la pelvis. La TC realzada con contraste tiene sensibilidad y especificidad altas para la detección de trombos tumorales cavoatriales en pacientes con tumor de Wilms, lo que quizás afecte el abordaje quirúrgico. Es posible que la evaluación Doppler de rutina se realice después de la TC, pero no es necesaria.[164] Si el tumor se encuentra en las venas hepáticas o por encima de ellas, se sugiere una biopsia con quimioterapia preoperatoria debido a la tasa más baja de complicaciones intraoperatorias graves. Se deben controlar los trombos tumorales grandes antes del abordaje quirúrgico de la masa renal, en especial cuando los trombos se extienden por encima de la vena hepática, con el fin de evitar la embolización del tumor. En algunos casos, se requiere derivación cardiopulmonar.[177]
- A veces los tumores de Wilms se rompen antes de la cirugía. Los términos rotura y ruptura se utilizan para indicar que se rompió la cápsula del tumor antes de la cirugía, mientras que los términos derrame y desbordamiento se refieren a una rotura del tumor durante la cirugía. De acuerdo con desempeños diagnósticos semejantes, tanto la TC como la IRM son útiles para la detección de una rotura. Si bien los hallazgos de las imágenes compatibles con una rotura tienen una especificidad alta (88 %), el diagnóstico de la rotura se debe confirmar en la cirugía. No se pueden usar solo imágenes para la estadificación inicial debido a la sensibilidad y especificidad bajas para la rotura preoperatoria y el estado de los ganglios linfáticos.[167]
Pronóstico y factores pronósticos del tumor de Wilms
El tumor de Wilms es una enfermedad curable en la mayoría de los niños afectados. Desde la década de los 80, la tasa de supervivencia a 5 años del tumor de Wilms de tipo histológico favorable (HF) se ha mantenido constante por encima del 90 %.[178] Este desenlace favorable se produjo con cambios en el tratamiento que incluyeron reducciones en la duración del tratamiento, la dosis de radiación, la extensión de los campos irradiados y el porcentaje de pacientes que recibieron radioterapia.[179]
El pronóstico de los pacientes con tumor de Wilms depende de los siguientes aspectos:[180,181,182,183]
- Características histopatológicas del tumor (de tipo HF vs. de tipo histológico anaplásico). Para obtener más información, consultar la sección Hallazgos histológicos en el tumor de Wilms.
- Estadio de la enfermedad en el momento del diagnóstico.
- Características moleculares del tumor, como ganancia de 1q y pérdida de heterocigosidad de 1p y 16q. La ganancia de 1q, que afecta al 28 % de los tumores de Wilms, es el factor pronóstico más potente del desenlace y se relaciona con un resultado adverso.[106,107,109] La pérdida de heterocigosidad de 11p15 y la pérdida de impronta de 11p15 se relacionan con recaída en pacientes de riesgo muy bajo que no reciben quimioterapia.[109,184] Para obtener más información, consultar la sección Características genómicas del tumor de Wilms.
- Edad. La edad en el momento de la presentación suele oscilar entre 2 y 5 años, y la incidencia del tumor de Wilms en niños mayores de 10 años es escasa. La edad avanzada se relaciona con un pronóstico adverso.[185,186]
Adolescentes mayores y adultos con tumor de Wilms
El tumor de Wilms en pacientes mayores de 16 años es poco frecuente, con una tasa de incidencia de alrededor de 0,2 casos por millón por año en pacientes de 15 a 39 años.[2] La tasa relativa de supervivencia a 5 años es del 75 % para este grupo de pacientes.[2] En Europa, la mediana de edad en el momento del diagnóstico para los pacientes adultos con tumor de Wilms (definida como edad >15 años) es de 34 años. Sin embargo, se notificaron pacientes mayores de 60 años.[187] El 3 % de los tumores de Wilms se presentan en adultos. El tumor de Wilms representa menos del 1 % de todos los tumores renales en adultos y a veces es un hallazgo inesperado después de una nefrectomía por un supuesto carcinoma de células renales, que es el cáncer de riñón más común en adultos.
El tumor de Wilms que se presenta en los adultos difiere del que se presenta en los niños de varias maneras. Los adultos rara vez presentan enfermedad bilateral (<1 %). Más pacientes adultos presentaron neoplasias malignas primarias adicionales (antes y después del diagnóstico de tumor de Wilms) en comparación con sus contrapartes pediátricas.[188] No se ha observado que los tumores de Wilms que se presentan en adultos se relacionen con restos nefrogénicos ni con afecciones del desarrollo como los síndromes WAGR, de Denys-Drash o de Beckwith-Wiedemann.[135] Para obtener información sobre las características moleculares del tumor de Wilms en adultos, consultar la sección Características genómicas del tumor de Wilms.
Una situación específica para los adultos es el diagnóstico de tumor de Wilms en mujeres embarazadas. Este diagnóstico se realiza de manera incidental durante el seguimiento ecográfico del embarazo o debido a síntomas clínicos como dolor abdominal y fiebre.[189,190]
Los desenlaces de los adolescentes y adultos jóvenes (AAJ) (15 a 39 años) y los adultos son inferiores a los desenlaces de los niños.
- En un análisis de pacientes con tumor de Wilms en la base de datos Surveillance, Epidemiology, and End Results (SEER), los pacientes AAJ (n = 104) tuvieron una tasa de SG a 5 años estadísticamente inferior (69 vs. 94 %; P < 0,001) que los pacientes pediátricos (n = 2 574).[191][Nivel de evidencia C1]
El desenlace inferior en los adultos quizás sea multifactorial, y se explica por factores como las diferencias en las características biológicas tumorales entre niños y adultos, el diagnóstico incorrecto, la estadificación inadecuada (por ejemplo, es más probable que se estadifique como enfermedad localizada o que no obtengan muestras ganglionares), el tratamiento insuficiente o el cumplimiento terapéutico precario (por ejemplo, sin radioterapia), la falta de familiaridad de los oncólogos y patólogos con el tumor de Wilms en adultos (que a veces conduce a un diagnóstico erróneo o demorado), el retraso para iniciar el tratamiento apropiado adaptado al riesgo, así como la carencia de protocolos de tratamiento específicos para adultos.[192][Nivel de evidencia C1]
Tratamiento de adultos con tumor de Wilms
Dado que el tumor de Wilms rara vez se presenta en adultos, no hay un protocolo de tratamiento estándar. Se han notificado desenlaces más favorables en los adultos que reciben tratamiento en ensayos pediátricos.
Un tumor de Wilms en un adulto representa una emergencia terapéutica debido al crecimiento rápido del tumor y porque los urólogos y oncólogos están más familiarizados con el crecimiento lento del carcinoma de células renales.
El NWTS Group notificó los desenlaces para pacientes adultos con tumor de Wilms de los ensayos NWTS-1, 2 y 3.[193,194,195]
- La tasa de SG a 3 años para los adultos en el ensayo NWTS-1 fue del 24 % (en comparación con el 74 % en los niños) y mejoró hasta una tasa de SG a 5 años del 82,6 % en el ensayo NWTS-3, aunque el número de pacientes adultos tratados en cada ensayo fue de 31 o inferior.
- Estos datos indican que se anticipa la curación de muchos adultos con tumor de Wilms cuando su tratamiento es apropiado; en especial, si el tumor no se diseminó y se resecó por completo.
Para los adultos con enfermedad resistente al tratamiento o recidivante, se deben considerar los exámenes de detección de posibles dianas terapéuticas en el tumor.[196]
En las siguientes recomendaciones de los comités de tumores renales de la International Society of Pediatric Oncology (SIOP) y el COG, se promueve el uso de un abordaje uniforme para mejorar el desenlace de los adultos con tumor de Wilms.[197]
- Consultar a un oncólogo pediatra con experiencia en el tratamiento del tumor de Wilms tan pronto como se sospeche el diagnóstico histológico.
- Evitar la demora para comenzar la quimioterapia. En teoría, la quimioterapia y la radioterapia, si fueran necesarias, se deben iniciar el día 14 después de la nefrectomía, aunque se acepta la demora del inicio hasta el día 30.
- Vigilar la aparición de los efectos tóxicos de la vincristina (neurotoxicidad) y la dactinomicina (toxicidad hepática) en adultos.
- Inscribir a los pacientes en los ensayos clínicos de tumores renales pediátricos disponibles cuando los pacientes cumplen con los requisitos.
En una serie de 14 adultos con tumores de Wilms evaluados mediante secuenciación dirigida ampliada, en 5 (36 %) se encontraron variantes BRAF V600E. Estos tumores contenían áreas morfológicamente idénticas al adenoma metanéfrico con alteración BRAF V600E. Todos los tumores de Wilms con alteración BRAF V600E de esta cohorte se presentaron en pacientes mayores de 30 años. La identificación de una variante BRAF V600E tiene importancia terapéutica porque estos pacientes pueden responder al tratamiento con inhibidores de BRAF o MEK.[135] Hubo un informe de un hombre (51 años) que tenía un tumor de Wilms metastásico en recaída que albergaba una variante BRAF V600E. El paciente se trató con el inhibidor de BRAF dabrafenib y tuvo una respuesta prolongada e intensa.[196]
Hallazgos histológicos en el tumor de Wilms
Aunque la mayoría de los pacientes con un diagnóstico histológico de tumor de Wilms evolucionan bien con el tratamiento actual, cerca del 10 % de ellos exhiben características histopatológicas que se relacionan con un pronóstico más adverso y, en algunos tipos, con una incidencia alta de recaída y muerte. El tumor de Wilms se divide en los siguientes dos grupos pronósticos a partir de las características histopatológicas tumorales y renales:
Tipo histológico favorable
Desde el punto de vista histológico, el tumor de Wilms imita el desarrollo trifásico del riñón normal, que comprende tipos celulares blastémicos, epiteliales (tubulares) y estromales. No todos los tumores son trifásicos; los tumores monofásicos acarrean dificultades para el diagnóstico.
Si bien se indicó un vínculo entre las características histológicas y el pronóstico o el grado de respuesta al tratamiento, ninguna de estas características alcanzó significación estadística en los algoritmos de tratamiento de América del Norte, excepto la anaplasia. Por lo tanto, las características histológicas no dirigen el tratamiento inicial.[198] Se ha identificado una fuerte relación entre el subtipo epitelial y las variantes de TRIM28.[124][Nivel de evidencia C1]
En el estudio AREN03B2 (NCT00898365), se analizó a los pacientes con enfermedad en estadio I a partir de las características histológicas epiteliales (n = 177) y el tratamiento. Cuando se analizó por características histológicas epiteliales, la tasa de SSC a 4 años fue del 96 % y la tasa de SG fue del 100 %. Cuando se analizó a estos pacientes según el tratamiento, los pacientes tratados con vincristina y dactinomicina (régimen EE-4A) (n = 117) tuvieron una tasa de SSC a 4 años del 96 %, en comparación con una tasa de SSC a 4 años del 98 % en los pacientes sometidos a nefrectomía sola (n = 57) (P = 0,549).[199]
En una serie de 14 adultos (17 a 46 años) con tumores de Wilms, la secuenciación dirigida de tumores reveló variantes BRAF V600E en 5 de los tumores. Todos estos tumores tenían áreas mejor diferenciadas que eran idénticas al adenoma metanéfrico, en combinación con el tumor epitelial de Wilms. Los adultos que tienen tumores con esta manifestación histológica se pueden beneficiar de la secuenciación de sus tumores.[135]
Tipo histológico anaplásico
El tipo histológico anaplásico representa alrededor del 10 % de los casos de tumor de Wilms. Este tipo histológico anaplásico es el principal factor predictor del desenlace y la supervivencia en los pacientes con tumor de Wilms. Los tumores que se presentan en pacientes de más edad (10 a 16 años) tienen una incidencia más alta del tipo histológico anaplásico.[200] En los tumores bilaterales, se notificó que entre el 12 % y el 14 % presentan características histológicas anaplásicas en un riñón.[201,202]
Los dos criterios histológicos que deben estar presentes con el fin de confirmar el diagnóstico de anaplasia son los siguientes:
- Presencia de figuras mitóticas poliploides multipolares con agrandamiento nuclear marcado.
- Hipercromasia.
Los cambios en 17p congruentes con variantes del gen TP53 se relacionaron con focos del tipo histológico anaplásico.[119] La anaplasia focal se define como la presencia de una o más regiones de anaplasia muy localizadas en un tumor primario. Todas estos factores respaldan la hipótesis de que la anaplasia evoluciona como un episodio tardío de una subpoblación de células del tumor de Wilms que adquirieron más lesiones genómicas.[203] La anaplasia focal no confiere un pronóstico tan precario como la anaplasia difusa.[182,204,205]
La anaplasia se correlaciona mejor con el grado de respuesta al tratamiento que con el grado de malignidad tumoral. Se relaciona más con un pronóstico adverso cuando se distribuye de manera difusa y cuando se identifica en estadios avanzados. Estos tumores son más resistentes a la quimioterapia tradicional que se usa en niños con tumor de Wilms de tipo HF.[182]
Prueba germinal deTRIM28
Para identificar a los pacientes con variantes germinales de TRIM28, se debe realizar una evaluación sistemática de los tumores de Wilms mediante prueba inmunohistoquímica con el anticuerpo anti-KAP1 para detectar la pérdida de TRIM28. Aunque la mayoría de los tumores con alteración de TRIM28 son tumores de Wilms con predominio epitelial, se deben considerar las pruebas para todos los subtipos, porque se notificó que otros subtipos histológicos tienen la variante TRIM28. Posteriormente, se puede realizar un análisis genético de TRIM28 en el DNA derivado de la sangre en todos los pacientes que presentan pérdida de TRIM28 en el tumor.[127]
Restos nefrogénicos
Los restos nefrogénicos son grupos de células precursoras embrionarias del riñón retenidas de forma anormal (pasadas las 36 semanas) y organizadas en racimos. Se encuentran restos nefrogénicos en cerca del 1 % de las autopsias pediátricas no seleccionadas, el 35 % de los riñones de pacientes con tumor de Wilms unilateral y prácticamente en el 100 % de los riñones de pacientes con tumor de Wilms bilateral.[83,206] La quimioterapia preoperatoria no afecta la prevalencia general de los restos nefrogénicos. Se notificaron anomalías congénitas en el 12 % de los pacientes con restos nefrogénicos, incluso en el 9 % de los pacientes con tumor de Wilms unilateral y en el 33 % de los pacientes con enfermedad bilateral.[8]
El término nefroblastomatosis se define como la presencia de restos nefrogénicos difusos o multifocales. En los tumores de Wilms unilaterales, los restos nefrogénicos por lo general solo son detectables mediante análisis histológico, mientras que en los tumores bilateral, los restos nefrogénicos proliferantes a veces son lo suficientemente grandes como para observarse en las imágenes.[207] Los restos nefrogénicos se subclasifican según la ubicación anatómica de los restos (intralobulares o perilobulares), y la fase de crecimiento (incipientes o inactivos, hiperplásicos, y en regresión o esclerosantes). Los defectos genéticos subyacentes afectan la presencia de restos nefrogénicos.[8] Con frecuencia, los tumores de Wilms relacionados con WT1 tienen pocos restos nefrogénicos intralobulares ubicados en el centro, junto a la médula renal o adyacentes a esta. Los tumores de Wilms relacionados con TRIM28 o con el síndrome de Beckwith-Wiedemann tienden a albergar restos nefrogénicos perilobulares en el tejido renal adyacente. Aunque solo se han analizado unos pocos restos nefrogénicos, estos parecen albergar incluso menos variantes que los tumores de Wilms adyacentes.[208,209]
La diferenciación entre los restos nefrogénicos y el tumor de Wilms mediante imágenes es problemática porque hay una superposición de su apariencia. En un estudio retrospectivo, se evaluó a 52 niños pequeños (edad, <5 años) con restos nefrogénicos y tumores de Wilms pequeños (todas las lesiones <5 cm) para los que se había obtenido una muestra mediante cirugía y realizado el análisis patológico antes de cualquier intervención médica. Los investigadores encontraron que se debe preferir el diagnóstico de tumor de Wilms en lugar del de resto nefrogénico cuando la masa renal es esférica, exofítica y su diámetro máximo mide más de 1,75 cm. La homogeneidad en las imágenes favorece el diagnóstico de restos nefrogénicos perilobulares, mientras que la ausencia de homogeneidad es más probable en los restos intralobulares y en el tumor de Wilms.[165][Nivel de evidencia C1]
La nefroblastomatosis perilobular hiperplásica difusa es una categoría única de nefroblastomatosis en la que se forma una corteza gruesa alrededor de un riñón o de ambos riñones, y se considera una afección preneoplásica. La diferenciación entre el tumor de Wilms y los restos nefrogénicos perilobulares hiperplásicos difusos a veces es problemática; es esencial examinar la unión entre la lesión y el parénquima renal adyacente. Las biopsias por incisión no tienen utilidad diagnóstica a menos que incluyan el borde entre la lesión y el parénquima renal normal.[210]
El tipo y porcentaje de restos nefrogénicos varía en pacientes con enfermedad unilateral o bilateral. Los pacientes con tumor de Wilms bilateral tienen una proporción más alta de restos perilobulares (52 %) que de restos intralobulares o combinados (32 %), además de proporciones relativamente más altas de restos en comparación con los pacientes con tumor unilateral (18 % perilobulares y 20 % intralobulares, o ambos).[211] Los restos nefrogénicos intralobulares se relacionaron con el tumor de Wilms de tipo estromal y una edad más temprana en el momento del diagnóstico.[8]
Se considera que los pacientes con cualquier tipo de restos nefrogénicos en un riñón extirpado por un nefroblastoma tienen un riesgo más alto de formación de un tumor en el riñón que queda. Este riesgo disminuye con la edad del paciente.[212]
Para obtener información sobre el tratamiento de la nefroblastomatosis perilobular hiperplásica difusa bilateral, consultar la sección Nefroblastomatosis.
Los restos nefrogénicos extrarrenales son poco frecuentes y en ocasiones se convierten en tumor de Wilms extrarrenal.[213]
Información sobre los estadios del tumor de Wilms
El estadio se determina de acuerdo con los resultados de los estudios con imágenes, además de los hallazgos quirúrgicos y patológicos de la nefrectomía. El estadio es el mismo para los tumores de tipo HF y de tipo anaplásico. Por lo tanto, la información sobre el estadio se caracteriza por una declaración que incluye ambos criterios (por ejemplo, estadio II, de tipo HF o estadio II, de tipo histológico anaplásico).[198,214]
En un principio, el grupo NWTS formuló el sistema de estadificación que el COG todavía utiliza. A continuación se expone el sistema de estadificación que se utiliza en América del Norte y la incidencia por estadio.[198] Con el fin de obtener el estadio más exacto, es muy recomendable el uso del muestreo de ganglios linfáticos en todos los pacientes, aunque no tengan ganglios clínicamente anormales.
Estadio I
En el tumor de Wilms en estadio I (43 % de los pacientes), se deben satisfacer todos los criterios siguientes:
- El tumor se limita al riñón y el tumor se resecó por completo.
- La cápsula renal está intacta.
- El tumor no se rompió ni se obtuvo una biopsia antes de la extirpación.
- No hay compromiso de los vasos del seno renal.
- No hay indicios de tumor en los márgenes de la resección ni fuera de estos márgenes.
- Todas las muestras de ganglios linfáticos dan negativo.
Para que un tumor sea apto para tratarse con ciertos protocolos terapéuticos, como los de estadio I de riesgo muy bajo, los ganglios linfáticos regionales se deben examinar al microscopio.
Estadio II
En el tumor de Wilms en estadio II (20 % de los pacientes), el tumor se resecó por completo y no hay indicios de tumor en los márgenes de la resección o fuera de estos márgenes. El tumor se extiende fuera del riñón como lo indica cualquiera de los criterios siguientes:
- Hay extensión regional del tumor (es decir, penetración de la cápsula renal o invasión extensa del tejido blando del seno renal, según se explica a continuación).
- En la pieza de la nefrectomía, los vasos sanguíneos fuera del parénquima renal, incluso los vasos del seno renal, contienen células tumorales. Los márgenes están libres de tumor.
- La extensión vascular del tumor solo se considera en estadio II si se extirpó por completo en bloque con la pieza de la nefrectomía.
Todas las muestras de ganglios linfáticos dan negativo.
El COG Renal Tumor Committee (COG RTC) incluye ahora la ruptura o derrame confinado al flanco, incluso la biopsia del tumor, en el estadio III; sin embargo, los datos que respaldan este abordaje son controvertidos.[172,215]
Estadio III
En el tumor de Wilms en estadio III (21 % de los pacientes), se encuentra tumor residual no hematógeno limitado al abdomen después de la cirugía. Es posible que se presente cualquiera de las siguientes situaciones:
- Compromiso tumoral de ganglios linfáticos en el abdomen o la pelvis. (El compromiso ganglionar en el tórax u otros sitios extraabdominales es un criterio del estadio IV).
- El tumor penetró la superficie peritoneal.
- Se encuentran implantes tumorales en la superficie peritoneal.
- Queda tumor macroscópico o microscópico después de la cirugía (por ejemplo, se encuentran células tumorales en el margen de la resección quirúrgica durante el examen microscópico).
- No es posible la extirpación completa del tumor debido a infiltración local de estructuras vitales.
- La rotura tumoral antes de la cirugía o cualquier derrame durante la cirugía se consideran criterios del estadio III.
- La biopsia se obtiene antes de extirpar el tumor (ya sea biopsia con aguja Tru-cut, biopsia abierta o biopsia por aspiración con aguja fina).
- Se extirpa el tumor en más de una pieza (por ejemplo, se encuentran células tumorales en una glándula suprarrenal que se extirpó por separado; se extrae un trombo tumoral de la vena renal en una pieza separada de la pieza de la nefrectomía). La extensión del tumor primario por la vena cava hacia la vena cava torácica y el corazón se consideran criterios del estadio III, en lugar del estadio IV. Aunque son sitios extraabdominales, y a veces se consideran criterios del estadio II si el tumor se extirpa por completo en bloque con la pieza de nefrectomía.
El compromiso ganglionar y la enfermedad residual microscópica se notifican como factores de predicción alta del desenlace en pacientes con tumor de Wilms de tipo HF en estadio III.[216]
Estadio IV
En el tumor de Wilms en estadio IV (11 % de los pacientes), se debe satisfacer uno de los criterios siguientes:
- Metástasis hematógenas (pulmonares, hepáticas, óseas y encefálicas).
- Metástasis ganglionares fuera de la región abdominopélvica.
La presencia de un tumor dentro de la glándula suprarrenal no se interpreta como una metástasis y la estadificación depende del resto de los parámetros de estadificación presentes. De acuerdo con los criterios mencionados antes, se asigna un estadio local para el tumor primario, lo que determina la terapia local. Por ejemplo, es posible que un paciente presente una enfermedad en estadio IV con estadio local III.
Estadio V (bilateral)
En el tumor de Wilms en estadio V (5 % de los pacientes), se encuentra compromiso tumoral bilateral en el momento del diagnóstico. En el paradigma vigente, todos los pacientes con tumor de Wilms bilateral reciben el mismo tratamiento durante las primeras 6 a 12 semanas. Después de la cirugía definitiva, el tratamiento se basa en el estadio más alto del tejido renal que queda y el análisis patológico después del tratamiento.[175]
Tratamiento del tumor de Wilms
Aspectos generales de las opciones de tratamiento del tumor de Wilms
Debido a que el tumor de Wilms suele ser relativamente raro, se debe considerar la inscripción en un ensayo clínico de todos los pacientes con tumor de Wilms. Para determinar y llevar a cabo el tratamiento óptimo, es necesario que su planificación y administración estén a cargo de un equipo multidisciplinario de especialistas en cáncer (cirujano pediatra o urólogo pediatra, radioncólogo pediatra y oncólogo pediatra) con experiencia en el tratamiento de niños con tumor de Wilms.
Abordajes del Children's Oncology Group y la International Society of Pediatric Oncology para el tratamiento del tumor de Wilms
La mayoría de los estudios clínicos aleatorizados sobre el tratamiento de niños con tumor de Wilms han estado a cargo de dos grandes grupos clínicos (COG RTC y SIOP). Las diferencias entre los dos grupos afectan la estadificación y la clasificación. Hay dos abordajes estándar para el tratamiento del tumor de Wilms: el COG RTC usa cirugía inmediata para todos los tumores unilaterales, y la SIOP usa quimioterapia preoperatoria como primer paso del tratamiento. Ambos grupos emplean quimioterapia posoperatoria, excepto en casos seleccionados que no reciben quimioterapia, y en los pacientes en estadio avanzado, en quienes se administra radioterapia como abordaje adaptado al riesgo.
- COG RTC (incluye el grupo NWTS anterior): el grupo NWTS estableció el tratamiento estándar para el tumor de Wilms en América del Norte, que consta de una nefrectomía inicial (cuando es posible) seguida de quimioterapia y, en algunos pacientes, radioterapia.[217,218,219] Este abordaje facilita un diagnóstico histológico temprano y exacto, la extracción de material biológico que no esté afectado por el tratamiento y la recogida de información de estadificación (como la presencia de derrame tumoral o compromiso tumoral de ganglios linfáticos), antes de administrar quimioterapia.
- SIOP: la SIOP es un consorcio europeo que en sus ensayos administra quimioterapia preoperatoria antes de la resección definitiva a los pacientes con tumores renales. Esto se traduce en menos derrames tumorales durante la cirugía y un estadio posoperatorio más bajo.[220] Cuando se compararon las características histológicas de los tumores de Wilms entre los pacientes sometidos a cirugía inmediata y los pacientes que recibieron quimioterapia preoperatoria, se observó que la quimioterapia preoperatoria alteró de forma significativa las características histológicas tumorales, y se encontraron menos casos de los tipos histológicos blastémicos y mixtos. Además, hubo menos tumores en estadio III en el grupo de quimioterapia preoperatoria.[221]
- Tanto la SIOP como el COG tratan a los lactantes menores de 6 meses de vida con una nefrectomía primaria.[222]
Este resumen se concentra en los estudios del NWTS (en la actualidad, COG RTC) y en los resultados de estos estudios.
Las principales conclusiones sobre el tratamiento y otros aspectos de los estudios NWTS-1, NWTS-2, NWTS-3, NWTS-4 y NWTS-5 son las siguientes:
- No es necesario el uso rutinario de radioterapia posoperatoria dirigida al flanco en niños con tumores de tipo HF en estadio I o estadio II cuando se administra quimioterapia combinada de vincristina y dactinomicina después de la nefrectomía.[219]
- El pronóstico de los pacientes con tumor de tipo HF en estadio III es mejor cuando el tratamiento incluye una de los siguientes opciones: a) dactinomicina, vincristina, doxorrubicina y 10,8 Gy de radioterapia dirigida al flanco; o b) dactinomicina, vincristina y 20 Gy de radioterapia dirigida al flanco. Se indica el uso de radiación dirigida a todo el abdomen para la enfermedad intraperitoneal extensa o el derrame tumoral intraperitoneal generalizado, y es posible que se administre un refuerzo para la enfermedad residual macroscópica.[219]
- La adición de ciclofosfamida en la dosis del protocolo (10 mg/kg/d por 3 días cada 6 semanas) a la combinación de vincristina, dactinomicina y doxorrubicina no mejora el pronóstico de los pacientes con tumores de tipo HF en estadio IV.[219]
- Una sola dosis de dactinomicina por curso (de tipo HF en estadio I o II, de tipo anaplásico en estadio I, de tipo HF en estadio III, estadios III o IV, o sarcoma de células claras de riñón en estadios I–IV), equivale a los cursos de dosis divididas, produce la misma SSC, logra mayor intensidad de la dosis y se relaciona con menos efectos tóxicos y gastos.[223]
- En los pacientes con un tumor de tipo HF en estadio I y estadio II es suficiente el tratamiento durante 18 semanas, y en los pacientes en estadio III y estadio IV es posible administrar tratamiento durante 6 meses en lugar de 15 meses.[179,217,223,224,225]
- La ganancia de 1q se relaciona con una supervivencia inferior en pacientes con tumor de Wilms unilateral de tipo HF. Este es el factor independiente más potente para predecir el desenlace; cuando hay ganancia de 1q, no son importantes las pérdidas de 1p o 16q. Cuando no hay ganancia de 1q en el tumor de Wilms unilateral de tipo HF, las pérdidas de 1p y 16q conservan parte de la importancia pronóstica y acarrean un riesgo más alto de recidiva.[106,109]
Cirugía
Los siguientes principios quirúrgicos también evolucionaron debido a los ensayos del NWTS (COG):
- La función más importante del cirujano es asegurar la extirpación completa del tumor sin rotura, y evaluar la extensión de la enfermedad. El procedimiento de elección es la nefrectomía radical y el muestreo de ganglios linfáticos a través de una incisión transabdominal o toracoabdominal.[226] No se realiza una incisión en el flanco porque proporciona una exposición limitada del riñón.
En los pacientes con tumores resecables no se obtienen biopsias preoperatorias ni durante la cirugía porque ambas suben la estadificación tumoral en el sistema del COG vigente.[226]
- No es necesario hacer una exploración de rutina del riñón contralateral si los estudios con imágenes técnicamente adecuadas no indican un proceso bilateral. Si los estudios con imágenes iniciales indican compromiso renal bilateral, los abordajes de tratamiento deben facilitar la cirugía con conservación renal.[163]
- Alrededor de 2 % de los casos de tumor de Wilms presentan compromiso ureteral. La presencia de hematuria macroscópica, un riñón no funcionante o hidronefrosis indican que quizás el tumor se diseminó dentro del uréter y se recomienda una cistoscopia. Se recomienda la resección en bloque para evitar el derrame tumoral.[227]
- El cirujano debe estar al tanto del riesgo de derrame intraoperatorio, en especial, en los pacientes con tumores grandes y del lado derecho, como se observó en una revisión de casos de derrame intraoperatorio en 1131 participantes en el estudio del COG AREN03B2 (NCT00898365).[228]
- Se debe considerar la resección del tumor renal aunque sea evidente una enfermedad en estadio IV en las pruebas con imágenes (por ejemplo, metástasis pulmonares). El tratamiento del tumor de Wilms en estadio I o II local, con metástasis a distancia no exige el uso de radioterapia local.
La cirugía con conservación del riñón continúa siendo polémica y los datos no la respaldan, salvo en niños que exhiban las siguientes características:[229,230]; [231][Nivel de evidencia C1]
- Un riñón único.
- Predisposición a tumores bilaterales. En algunos niños con predisposición a tumores bilaterales que tienen tumores muy pequeños identificados mediante exámenes de detección con ecografía se puede considerar el uso de una cirugía con conservación del tejido renal.[229]
- Riñón en forma de herradura. El tumor de Wilms que surge en un riñón en forma de herradura es raro, y es importante un diagnóstico preoperatorio exacto para planificar el abordaje operatorio. En la mayoría de los casos, es posible una resección primaria. Los casos inoperables por lo general se pueden resecar después de la quimioterapia.[232]
- Tumor de Wilms en lactantes con síndrome de Denys-Drash o síndrome de Frasier (para diferir la necesidad de diálisis).
La cirugía con conservación del riñón no es factible para la mayoría de los pacientes en el momento del diagnóstico debido a la ubicación del tumor dentro del riñón, incluso en los pacientes con tumores de riesgo muy bajo.[233] En América del Norte, la cirugía con conservación del riñón (nefrectomía parcial) de un tumor de Wilms unilateral después de la administración de quimioterapia para reducir la masa tumoral está en etapa de investigación.[234,235]
El tumor de Wilms no suele invadir los órganos adyacentes; en consecuencia, la resección de órganos contiguos solo se indica en contadas ocasiones. Hay una mayor incidencia de complicaciones en las resecciones más extensas que abarcan la extirpación de otros órganos además del diafragma y la glándula suprarrenal. Este hallazgo condujo a que en los protocolos del COG actuales, se recomiende que en aquellos pacientes en quienes se exige nefrectomía con extirpación de otros órganos, se considere obtener una biopsia inicial, administrar quimioterapia neoadyuvante y, luego, hacer una resección secundaria.[236] No se recomienda la resección primaria de metástasis hepáticas.[237]
Muestreo de ganglios linfáticos
El estado de los ganglios linfáticos es un factor pronóstico importante del desenlace a largo plazo en pacientes con tumor de Wilms.[226] Los datos de los estudios sobre tumores de Wilms indican que el número y la ubicación de los ganglios linfáticos pueden afectar el tratamiento y el desenlace, aunque los protocolos de tumores renales del NWTS y COG nunca han definido el número de ganglios linfáticos o las ubicaciones de los ganglios linfáticos que se deben muestrear.[226] Se desconoce el número ideal de ganglios linfáticos que se deben muestrear.
- La tasa de SG a 5 años fue más baja en los pacientes con 0 ganglios linfáticos muestreados (87 vs. 91 % para 1–5 ganglios linfáticos, 93 % para 6–10 ganglios linfáticos, 95 % para >10 ganglios linfáticos; P = 0,005). En el análisis multivariante se confirmó una ventaja de supervivencia en los pacientes con 1 a 5 ganglios linfáticos muestreados (CRI, 0,6; P = 0,016), 6 a 10 ganglios linfáticos muestreados (CRI, 0,521 P = 0,048) y >10 ganglios linfáticos muestreados (CRI, 0,403; P = 0,039), en comparación con los pacientes con 0 ganglios linfáticos examinados.[238]
- En el NWTS-5, el fracaso en la toma de muestras de ganglios linfáticos se relacionó con un riesgo más alto de recaída en los pacientes en estadio I y II.[239] En el AREN0532 se observaron resultados similares con una SSC más precaria en los pacientes que no se sometieron a una biopsia de ganglio linfático. La SSC en los pacientes en estadio III sin muestreo de ganglios linfáticos fue del 84 % (n = 148), en comparación con el 89 % en aquellos pacientes con muestreo de ganglios linfáticos (n = 387) (P = 0,067).[240]
- El muestreo de ganglios linfáticos hiliares y periaórticos es apropiado incluso si los ganglios parecen normales.[226,238] Además, se debe tomar una muestra de cualquier cuenca ganglionar sospechosa. Los márgenes de la resección, el tumor residual y cualquier cuenca ganglionar sospechosa se deben marcar con grapas de titanio.
Derrames pleurales
La presencia de un derrame pleural no exigen cambio en el tratamiento. En una revisión retrospectiva multinstitucional de 1259 niños con tumor de Wilms recién diagnosticado, 94 (7,5 %) presentaron derrame pleural.[168][Nivel de evidencia C1]
- Los pacientes con derrame pleural eran mayores que aquellos sin derrame pleural (mediana, 4,3 vs. 3,5 años; P = 0,004) y tenían más probabilidad de presentar enfermedad en estadio avanzado (estadio local III, 83 vs. 51,6 %; P = 0,0001).
- Solo 14 pacientes se sometieron a toracocentesis, y 3 de ellos tenían enfermedad metastásica parenquimatosa pulmonar en el momento del diagnóstico.
- De los pacientes con derrame pleural, 30 recibieron radioterapia torácica como parte de su tratamiento, y 29 de ellos tenían metástasis pulmonares o mediastínicas.
- Sesenta y cuatro pacientes con derrame pleural no recibieron radioterapia torácica, de los cuales 59 no tenían enfermedad pulmonar relacionada en el momento del diagnóstico.
- Solo 3 pacientes con derrame pleural y sin antecedentes de enfermedad pulmonar presentaron una recaída con enfermedad torácica.
- En toda la cohorte de pacientes con derrames pleurales, la tasa de SSC fue del 86,2 % y la tasa de SG fue del 91,5 %.
Quimioterapia
La quimioterapia preoperatoria antes de la nefrectomía se indica en las siguientes situaciones, que se han enumerado antes como situaciones que requieren una biopsia:[226,236,241,242,243,244]
- Tumor de Wilms en un riñón único.
- Tumor de Wilms bilateral sincrónico.
- Extensión de un trombo tumoral en la vena cava inferior por encima del nivel de las venas hepáticas. Casi el 4 % de los pacientes con tumor de Wilms presentan compromiso de la vena cava inferior o de las aurículas, y el 11 % presentan compromiso de la vena renal. La embolización de un trombo desde la vena cava hasta la arteria pulmonar es poco frecuente, pero puede causar la muerte; los trombos se deben identificar antes de la cirugía con el fin de prevenir la embolización y guiar el tratamiento.[164,177]
- Tumor que compromete estructuras contiguas de manera que la única forma de extirpar el tumor renal es extirpando otras estructuras (por ejemplo, bazo, páncreas o colon, pero no la glándula suprarrenal).
- Tumor de Wilms inoperable.
- Compromiso pulmonar por metástasis pulmonares extensas.
Para obtener más información, consultar la sección Evaluación diagnóstica y estadificación del tumor de Wilms.
En una serie contemporánea numerosa se incluyeron 124 pacientes con tumor de Wilms y extensión dentro de la cava tratados en centros de Norteamérica. La mayoría de los pacientes (82 %) recibió un régimen de quimioterapia neoadyuvante de 3 fármacos. Este tipo de quimioterapia reduce la necesidad de derivación cardiopulmonar y también evita la resección de trombos en la cava intrahepática, que resulta compleja.[244]
- En general, la tasa de regresión de los trombos fue del 45 %; la mejor respuesta se observó en el nivel suprahepático (62 %).
- Se evitó, potencialmente, la derivación cardiopulmonar en el 67 % de los pacientes.
- La tasa de complicaciones perioperatorias fue significativamente inferior después de la quimioterapia neoadyuvante (25 %), en comparación con la cirugía inicial (55 %; P = 0,005).
- En general, la tasa de SSC a 2 años fue del 93 %, y la tasa de SG fue del 96 %. Las tasas fueron más altas para los pacientes con tumores de tipo HF (98 % para HF vs. 82 % para tumores de tipo histológico desfavorable o anaplásico).
- La resección incompleta y la presencia de células viables en el trombo no afectó la SSC ni la SG.
Después de una biopsia se administra quimioterapia preoperatoria. La biopsia se puede realizar mediante un abordaje por el flanco.[177,245,246,247,248,249] Es imprescindible obtener tejido adecuado para la evaluación histológica exacta y los estudios moleculares. La quimioterapia preoperatoria incluye doxorrubicina además de vincristina y dactinomicina, a menos que se presenten características histológicas anaplásicas. En estos casos, la quimioterapia incluye el tratamiento con el régimen I (ver el Cuadro 2). Por lo general, la quimioterapia facilita la extirpación al disminuir el tamaño y el suministro vascular del tumor. Es posible que la quimioterapia también reduzca la frecuencia de las complicaciones quirúrgicas.[172,177,236,241,250,251]
En un metanálisis se investigó el efecto de la quimioterapia neoadyuvante en la viabilidad de un trombo del tumor de Wilms, donde la extensión intravascular se definió como todo tumor de Wilms con extensión más allá de la vena renal. Se encontró que la quimioterapia neoadyuvante fue eficaz para lograr la inviabilidad del trombo en alrededor del 50 % de los pacientes con tumor que invade la vena cava inferior. No se identificó ningún beneficio adicional con los ciclos prolongados de quimioterapia neoadyuvante. La mayoría de los pacientes recibieron quimioterapia con dactinomicina y vincristina con doxorrubicina o sin esta.[252]
En América del Norte, se ha sugerido el uso de quimioterapia preoperatoria en pacientes con indicios de una ruptura preoperatoria contenida para evitar el derrame intraoperatorio, pero esto es polémico.[253,254] El diagnóstico preoperatorio de una ruptura retroperitoneal contenida en la TC es difícil, incluso para radiólogos pediátricos experimentados.[161]
Todos los lactantes menores de 12 meses (incluso neonatos) que se tratarán con quimioterapia necesitan una reducción del 50 % de las dosis administradas a niños más grandes.[255] La dosificación para los lactantes (menores de 12 meses) se calcula por kilogramo de peso, no por área de superficie corporal. Esta reducción disminuye los efectos tóxicos notificados en los niños de este grupo de edad inscritos en estudios del NWTS, al tiempo que mantiene un resultado general excelente.[256]
Se debe controlar de cerca las pruebas del funcionamiento hepático en los niños con tumor de Wilms durante el curso inicial del tratamiento por la hepatotoxicidad grave (incluso síndrome de obstrucción sinusoidal, llamado antes enfermedad venooclusiva) notificada en estos pacientes.[257,258] En una cohorte de 8862 niños con tumores renales de los ensayos NWTS-3, 4 y 5, la incidencia de hepatopatía grave fue baja (0,8 %). La reintroducción cuidadosa de quimioterapia fue posible en la mayoría de los pacientes que presentaron efectos tóxicos graves en el hígado causados por la quimioterapia y la radioterapia.[259] No se debe administrar dactinomicina ni doxorrubicina durante la radioterapia.
Los pacientes que presentan insuficiencia renal durante el tratamiento pueden continuar con la quimioterapia de vincristina, dactinomicina y doxorrubicina. La vincristina y la doxorrubicina se pueden administrar en dosis completas. Sin embargo, la dactinomicina se relaciona con neutropenia grave. Es posible que no sea necesario reducir la dosis de estos fármacos, pero se necesitan estudios farmacológicos y farmacocinéticos precisos durante el tratamiento del paciente.[260,261]
El aumento del tratamiento mejora la SSC en los pacientes con tumor de Wilms de tipo HF y pérdida de heterocigosidad de 1p/16q. En los ensayos AREN0532 (NCT00352534) y AREN0533 (NCT00379340), pacientes con tumor de Wilms de tipo FH en estadio I y estadio II tratados con el régimen DD-4A (dactinomicina, vincristina, y doxorrubicina) demostraron una tasa de SSC a 4 años del 87,3 %, en comparación con una tasa de SSC a 4 años del 68,8 % (P = 0,042) en los pacientes en estadio I y estadio II tratados en el ensayo NWTS-5. Los pacientes con enfermedad en estadios III y IV presentaron una tasa de SSC a 4 años del 90,2 % cuando recibieron el régimen M (consultar el Cuadro 2), en comparación con el 61,3 % (P = 0,001) en los pacientes en estadio III y IV tratados en el ensayo NWTS-5. Se observó una tendencia hacia la mejora en las tasas de SG a 4 años en los pacientes en estadios I y II, y en los pacientes en estadios III y IV.[262][Nivel de evidencia C2]
Se exige el uso de radioterapia posoperatoria dirigida al lecho tumoral cuando se obtiene una biopsia o en el caso de un tumor localizado en estadio III. En un estudio de 1488 pacientes con tumores de Wilms sometidos a cirugía y radioterapia, el retraso en el inicio de la radioterapia después de la cirugía de más de 14 días se relacionó con un aumento del riesgo de mortalidad en los pacientes con tumor de Wilms no metastásico.[263][Nivel de evidencia C1]
En el Cuadro 2 se describen los regímenes quimioterapéuticos usados para el tratamiento del tumor de Wilms.
| Nombre del régimen | Descripción del régimen |
|---|---|
| Régimen EE-4A[109] | Vincristina y dactinomicina durante 18 semanas después de la nefrectomía |
| Régimen DD-4A[109] | Vincristina, dactinomicina y doxorrubicina durante 24 semanas; nefrectomía inicial o biopsia seguida de nefrectomía |
| Régimen I[182] | Vincristina, doxorrubicina, ciclofosfamida y etopósido durante 24 semanas después de la nefrectomía |
| Régimen M[264] | Vincristina, dactinomicina, doxorrubicina, ciclofosfamida y etopósido seguido de radioterapia |
| Régimen UH1[265] | Vincristina, doxorrubicina, ciclofosfamida, carboplatino y etopósido durante 30 semanas y radioterapia |
| Régimen UH2[265] | Vincristina, doxorrubicina, ciclofosfamida, carboplatino, etopósido, vincristina e irinotecán durante 36 semanas y radioterapia |
Radioterapia
La radioterapia se usa para mejorar el control local y tratar los sitios de enfermedad metastásica. Tradicionalmente la radioterapia dependía del estadio y el tipo histológico, pero en los últimos tiempos también se guía según la firma molecular del tumor.[184]
Abordaje del COG
La cirugía inicial permite obtener una confirmación histológica y determinar la extensión del tumor, lo que brinda la justificación para la terapia adyuvante, incluso la radioterapia. Además de los factores histológicos, los factores de riesgo posoperatorios para un control local más precario son: 1) resección incompleta, 2) márgenes positivos y 3) compromiso ganglionar. No se usa la radioterapia en los pacientes con tumor de Wilms de tipo HF en estadio I o II. En los pacientes con tumor de Wilms de tipo HF en estadio III, se usa la radioterapia dirigida al flanco o al abdomen como parte del tratamiento. En pacientes con un tipo histológico desfavorable (anaplasia focal o difusa), se indica la radioterapia dirigida al flanco o al abdomen en todos los casos. Para obtener más información, consultar el Cuadro 3.
Los resultados de los ensayos del NWTS (COG RTS) comprenden los siguientes aspectos:
- La radioterapia dirigida al flanco cubre el lecho tumoral, la región ganglionar comprometida y todos los cuerpos vertebrales adyacentes con una dosis de 10,9 Gy en fracciones de 1,8 Gy. La dosis de radioterapia se basa en los resultados del estudio NWTS-3 en el que no aumentó la recaída abdominal en los pacientes con tumor de tipo HF en estadio III que recibieron 10 Gy versus 20 Gy con la quimioterapia DD-4A.[266]
- La radioterapia dirigida a todo el abdomen comprende la administración de una dosis de 10,5 Gy en fracciones de 1,5 Gy y se usa para el tratamiento del derrame difuso o las metástasis peritoneales.[262]
- En el estudio del COG AREN0321 (NCT00335556), que ya se completó, la dosis de radioterapia dirigida al lecho tumoral fue de 10,8 Gy en fracciones de 1,8 Gy, excepto para los pacientes con anaplasia difusa en estadio III, que recibieron una dosis de 19,8 Gy en fracciones de 1,8 Gy. Este sigue siendo el estándar de tratamiento actual.[265]
- Los resultados de los primeros estudios del NWTS (1 y 2) indicaron que un retraso de la radioterapia de más de 10 días después de la cirugía produjo un control local más precario, en particular, en el tumor de Wilms de tipo histológico desfavorable.[267,268] Sin embargo, no se encontró diferencia en el control local cuando la administración de radioterapia se retrasó más de 10 días después de la cirugía en los pacientes con tumores de tipo HF en estadios II a IV tratados en los ensayos NWTS-3 y NWTS-4.[102] Los datos más recientes de la National Cancer Database confirmaron una mejora de la supervivencia en pacientes con tumor de Wilms no metastásico que recibieron radioterapia adyuvante 14 días o menos después de la cirugía.[263]
- En los resultados de los ensayos NWTS-3 y NWTS-4 se indica que no hay beneficio de supervivencia cuando se usa irradiación dirigida a todo el pulmón en el entorno de metástasis pulmonares identificadas solo en la TC.[269] Las pautas del COG actuales permiten omitir la irradiación dirigida a todo el pulmón en casos con enfermedad de tipo HF sin metástasis extrapulmonares, pérdida de heterocigosidad de 1p y 16q, y remisión completa a las 6 semanas de la administración de vincristina, dactinomicina y doxorrubicina.[184] Cuando se irradia todo el pulmón, se indica una dosis de 12 Gy en fracciones de 1,5 Gy para niños mayores de 12 meses, y una dosis de 10,5 Gy en fracciones de 1,5 Gy para pacientes menores de 12 meses con metástasis pulmonar.
- Otros sitios de enfermedad metastásica en el tumor de Wilms son infrecuentes, entre ellos, el hígado, los ganglios extraabdominales, el encéfalo y los huesos. En el estudio del COG AREN0533 (NCT00379340) las dosis de radioterapia en los pacientes menores de 16 años fueron de 19,8 Gy en fracciones de 1,8 Gy dirigidas al hígado y los ganglios residuales macroscópicos; de 21,6 Gy en fracciones de 1,8 Gy dirigidas a todo el encéfalo con un refuerzo de 10,8 Gy en fracciones de 1,8-Gy dirigidas a los sitios de enfermedad metastásica macroscópica en el encéfalo; y de 25,2 Gy en 14 fracciones para las metástasis óseas. En los pacientes mayores de 16 años, la dosis de radioterapia dirigida a todo el encéfalo y los huesos se aumenta hasta 30,6 Gy en fracciones de 1,8 Gy.
| Enfermedad local o locorregional | ||||
|---|---|---|---|---|
| RT = radioterapia. | ||||
| a Exige RT dirigida a todo el abdomen en fracciones diarias de 1,5 Gy. Los pacientes con implantes peritoneales difusos irresecables reciben 21 Gy. | ||||
| b Se administra irradiación dirigida a todo el pulmón en fracciones diarias de 1,5 Gy. | ||||
| c No todos los pacientes reciben radioterapia. | ||||
| d Se administra un refuerzo para la enfermedad macroscópica. | ||||
| Estadio I | Estadio II | Estadio III | Estadio III (derrame difuso, metástasis peritoneal, rotura preoperatoria)a | |
| Tipo histológico favorable | Sin RT | Sin RT | 10,8 Gy | 10,5 Gy |
| Tipo histológico de anaplasia local | 10,8 Gy | 10,8 Gy | 10,8 Gy | 10,5 Gy |
| Tipo histológico de anaplasia difusa | 10,8 Gy | 10,8 Gy | 19,8 Gy | 10,5 Gy y refuerzo de 9 Gy |
| Enfermedad metastásica | ||||
| Pulmonar, en estadio IV | Hepática, en estadio IV | Encefálica, en estadio IV | Ósea, en estadio IV | |
| Tipo histológico favorable | 10,5 Gy para la edad <12 mesesb,c; 12 Gy para la edad >12 mesesb,c | 19,8 Gy con refuerzo de 5,4 Gy a 10,8 Gy, o sin refuerzod | 21,6 Gy y refuerzo de 10,8 Gy en pacientes <16 años; 30,6 Gy en pacientes >16 años | 25,2 Gy en pacientes <16 años; 30,6 Gy en pacientes >16 años |
| Tipo histológico de anaplasia focal o difusa | 10,5 Gy para la edad <12 mesesb; 12 Gy para la edad >12 mesesb | 19,8 Gy con refuerzo de 5,4 Gy a 10,8 Gy, o sin refuerzod | 21,6 Gy y refuerzo de 10,8 Gy en pacientes <16 años; 30,6 Gy en pacientes >16 años | 25,2 Gy en pacientes <16 años; 30,6 Gy en pacientes >16 años |
Abordaje de la SIOP
A partir de la experiencia obtenida de los ensayos previos de la SIOP, los niños que necesitan radioterapia reciben tratamiento posoperatorio dirigido al flanco o a los sitios metastásicos. En los ensayos 1 a 9 de la SIOP se demostró que el uso preoperatorio de radioterapia o quimioterapia disminuyó la proporción de pacientes que presentaron derrame tumoral, desde más del 20 % hasta el 5 %. La ausencia de inferioridad de la quimioterapia preoperatoria en comparación con la radioterapia preoperatoria en el ensayo 5 de la SIOP, y la preocupación en torno a segundas neoplasias malignas cuando se usa radioterapia preoperatoria, llevó a la SIOP a recomendar la quimioterapia preoperatoria como el tratamiento inicial estándar.[220] Con el tiempo, el porcentaje de niños tratados con radioterapia posoperatoria disminuyó de más del 90 % al 15 % y al 25 % en los ensayos SIOP 6 a 9, SIOP 93-01 y SIOP-2001, respectivamente.[218]
Se omitió la radioterapia dirigida al abdomen en pacientes con tumor de Wilms metastásico local en estadio III que presentaron necrosis completa después de 6 semanas de quimioterapia preoperatoria. También se omitió en pacientes con tumor de Wilms en estadio III que recibieron 4 semanas de quimioterapia preoperatoria (n = 19) y presentaron necrosis completa. Los resultados fueron excelentes en los dos grupos de pacientes con tumor de Wilms en estadio III que presentaron necrosis completa. Las tasas de SSC y SG a 5 años fueron del 100 % en los pacientes con enfermedad en estadio III y del 95 % en los pacientes con enfermedad metastásica local en estadio III.[270,271]
Tratamiento del tumor de Wilms en estadio I
En el Cuadro 4, se ofrece una visión general de las opciones de tratamiento estándar y los datos de supervivencia de pacientes con tumor de Wilms en estadio I, a partir de resultados publicados.
| Tipo histológico | SSR o SSC a 4 años | SG a 4 años | Tratamientob |
|---|---|---|---|
| AD = tipo histológico anaplásico difuso; SSC = supervivencia sin complicaciones; AF = tipo histológico anaplásico focal; HF = tipo histológico favorable; LOH = pérdida de heterocigosidad; SG = supervivencia general; SSR = supervivencia sin recaída; RT = radioterapia. | |||
| a Fuente: Grundy et al.,[109]Shamberger et al.,[183]Fernandez et al.,[184]Dix et al.,[262]y Daw et al.[272] | |||
| b Para las descripciones del régimen de quimioterapia, consultar elCuadro 2. | |||
| c Un paciente presentó una recaída pulmonar 4,12 años después del diagnóstico. | |||
| HF <24 meses y peso tumoral <550 g | 90 % | 100 % | Cirugía, incluso biopsia de ganglio linfático sola |
| HF >24 meses y peso tumoral >550 g | SSR 94 % | 98 % | Nefrectomía con muestreo de ganglios linfáticos seguido del régimen EE-4A |
| HF con LOH 1p/16q (n = 8) | SSC 100 % | 100 % | Nefrectomía con muestreo de ganglios linfáticos seguido del régimen DD-4A |
| AF | 100 % | 100 % (n = 8) | Nefrectomía con muestreo de ganglios linfáticos seguidos del régimen EE-4A y RT |
| AD | 100 %c | 100 % (n = 10) | Nefrectomía con muestreo de ganglios linfáticos seguidos del régimen EE-4A y RT |
Evidencia (cirugía sola para los niños menores de 2 años en el momento del diagnóstico de un tumor de Wilms de tipo HF en estadio I con peso tumoral <550 g):
En el ensayo AREN0532 (NCT00352534), el COG validó los hallazgos del ensayo NWTS-5 que indican que la nefrectomía sola es el tratamiento adecuado para pacientes menores de 2 años en el momento del diagnóstico del tumor de Wilms de tipo HF en estadio I con un peso tumoral menor a 550 g.
- El ensayo AREN0532 (NCT00352534) se diseñó para confirmar los hallazgos del ensayo NWTS-5 sobre la posibilidad de prescindir de la quimioterapia adyuvante en niños menores de 2 años en el momento del diagnóstico de un tumor de Wilms de tipo HF en estadio I con peso tumoral inferior a 550 g. En este estudio se inscribieron 116 pacientes que reunieron los criterios de tumor de Wilms de riesgo muy bajo.[183,184,273]
- Se observó recidiva en 12 pacientes.
- La tasa de SSC a 4 años fue del 89,7 %, y la tasa de SG fue del 100 %.
- El estado de metilación de 11p15 se relacionó con recidiva (20 % de recidiva con pérdida de heterocigosidad, 25 % de recidiva con pérdida de la impronta y 3,3 % de recidiva con retención de la impronta normal [P = 0,011]).
- El riesgo de presentar tumor de Wilms metacrónico es muy bajo en los pacientes que tienen tumor de Wilms de riesgo muy bajo y que no exhiben indicios de un síndrome subyacente.
Evidencia (tratamiento del tumor de Wilms de tipo HF con predominio epitelial en estadio I):
- El COG notificó los desenlaces de pacientes de todas las edades con tumor de Wilms de tipo HF en estadio I que exhibían predominio epitelial. Cerca del 20 % de los tumores de Wilms de tipo HF en estadio I registrados en el AREN03B2 exhibieron predominio epitelial. En este grupo de 177 pacientes con tumores de Wilms con predominio epitelial en estadio I, 117 pacientes se trataron con EE-4A y 57 pacientes se clasificaron como tumor de Wilms de riesgo muy bajo y recibieron solo observación.[199][Nivel de evidencia C1]
- La tasa de SSC a 4 años fue del 96,2 % y la tasa de SG fue del 100 %.
- No hubo diferencia estadística en la SSC ni en la SG según la edad en el momento del diagnóstico (<48 meses y >48 meses) o el tratamiento (EE4A vs. observación sola).
- Se presentaron 6 episodios: 3 pacientes presentaron tumores contralaterales después del diagnóstico inicial y 2 de ellos habían recibido quimioterapia adyuvante para los tumores iniciales. Además, 3 pacientes presentaron enfermedad metastásica, y todos habían recibido EE4A como tratamiento primario.
Evidencia (tratamiento del tumor de Wilms anaplásico en estadio I):
- En el estudio AREN0321 (NCT00335556) se demostró que los desenlaces de los pacientes con tumor de Wilms anaplásico en estadio I mejoraron con la adición de doxorrubicina y radioterapia dirigida al flanco al régimen de vincristina y dactinomicina.[272]
- Las tasas de SSC y SG a 4 años estimadas fueron del 100 % en AREN0321, en comparación con el 70 % y el 81,5 %, respectivamente, en un análisis actualizado de 27 pacientes del NWTS-5 (mediana de seguimiento, 13,3 años). Un paciente con anaplasia difusa tuvo una recaída 4,12 años después del diagnóstico en el ensayo AREN0321.
- La adición de doxorrubicina y radioterapia en el ensayo AREN0321 se basó en las características de las recaídas abdominales y en sitios a distancia del tumor de Wilms anaplásico en estadio I que se observaron en el ensayo del NWTS-5.
- En el análisis retrospectivo de todos los pacientes con tumor de Wilms anaplásico en estadio I tratados en los ensayos NWTS-1 a NWTS-5 y AREN0321 se observó una mejora significativa en la SSC de los pacientes tratados con doxorrubicina (tasa de SSC a 4 años, 97,2 vs. 77,5 %; P = 0,01), pero no se observaron diferencias en la SSC según el uso de radioterapia dirigida al flanco (tasa de SSC a 4 años, 91,7 % vs. 80,2 %; P = 0,15).
- La tasa de recidiva local fue baja (3,6 %) y similar en los pacientes que recibieron radioterapia dirigida al flanco (4 %) y quienes no la recibieron (6,2 %). La recaída local ocurrió solo en pacientes con anaplasia difusa.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Tratamiento del tumor de Wilms en estadio II
En el Cuadro 5, se ofrece una visión general de las opciones de tratamiento estándar y los datos de supervivencia de pacientes con tumor de Wilms en estadio II, a partir de resultados publicados.
| Tipo histológico | SSR o SSC a 4 años | SG a 4 años | Tratamientob |
|---|---|---|---|
| AD = tipo histológico anaplásico difuso; SSC = supervivencia sin complicaciones; AF = tipo histológico anaplásico focal; HF = tipo histológico favorable; LOH = pérdida de heterocigosidad; SG = supervivencia general; SSR = supervivencia sin recaída; RT = radioterapia. | |||
| a Fuente: Grundy et al.,[109]Dome et al.,[182]Dix et al.,[262]y Daw et al.[265] | |||
| b Para las descripciones del régimen de quimioterapia, consultar elCuadro 2. | |||
| HF | SSR 86 % | 98 % | Nefrectomía con muestreo de ganglios linfáticos seguido del régimen EE-4A |
| HF con LOH 1p/16q (n = 24) | SSC 83 % | 100 % | Nefrectomía con muestreo de ganglios linfáticos seguido del régimen DD-4A |
| AF | SSC 80 % | 80 % (n = 5) | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| AD | SSC 84 % | 84 % (n = 19) | Nefrectomía con muestreo de ganglios linfáticos seguidos de RT abdominal y régimen UH1 |
Tratamiento de los pacientes con derrame intraoperatorio en estadio II
En una revisión de un grupo de 499 pacientes del ensayo NWTS-4 que tenían tumor de Wilms de tipo HF en estadio II, 95 presentaron un derrame tumoral. Las tasas de SSR a 8 años y de SG de los pacientes con derrame tumoral intraoperatorio tratados con vincristina y dactinomicina sin radioterapia dirigida al flanco fueron inferiores (75,7 % y 90,3 %, respectivamente) a las tasas de aquellos que no tenían un derrame tumoral (85 % y el 95,6 %, respectivamente). Ninguna de estas diferencias alcanzó la significación estadística.[215]
En los ensayos NWTS-3, NWTS-4 y NWTS-5, los pacientes con derrame intraoperatorio se dividieron en 2 grupos: 1) aquellos con derrame difuso que afectaba toda la cavidad abdominal y 2) aquellos con derrame local limitado al flanco. Los pacientes con derrame difuso recibieron radioterapia dirigida a todo el abdomen y quimioterapia de 3 fármacos (vincristina, dactinomicina y doxorrubicina); los pacientes con derrame local solo recibieron vincristina y dactinomicina. En los estudios del COG los pacientes con derrame local reciben tratamiento con doxorrubicina y radiación dirigida al flanco a partir de un análisis de los pacientes tratados en los estudios NWTS-3 y NWTS-4, en el que se observó una SG inferior en los pacientes con enfermedad en estadio II y derrame local en comparación con los pacientes sin derrame local.[274] Este abordaje es polémico y no se ha probado; por lo tanto, no se debe considerar estándar.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Tratamiento del tumor de Wilms en estadio III
En el Cuadro 6, se ofrece una visión general de las opciones de tratamiento estándar y los datos de supervivencia de pacientes con tumor de Wilms en estadio III, a partir de resultados publicados.
Para obtener más información sobre los pacientes clasificados en estadio III solo por la presencia de derrame local, consultar la sección Tratamiento del tumor de Wilms en estadio II.
| Tipo histológico | SSR o SSC a 4 años | SG a 4 años | Tratamientob |
|---|---|---|---|
| AD = tipo histológico anaplásico difuso; SSC = supervivencia sin complicaciones; AF = tipo histológico anaplásico focal; HF = tipo histológico favorable; LOH = pérdida de heterocigosidad; SG = supervivencia general; SSR = supervivencia sin recaída; RT = radioterapia. | |||
| A Fuente: Grundy et al.,[109]Dome et al.,[182]Fernandez et al.,[240]Dix et al.,[262]Y Daw et al.[265] | |||
| b Para las descripciones del régimen de quimioterapia, consultar elCuadro 2. | |||
| HF (todos los pacientes) | SSC 88 % | 97 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (sin LOH de 1p o 16q) y compromiso ganglionar (n = 109) | SSC 82 % | 97 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (sin LOH de 1p o 16q) y sin compromiso ganglionar (n = 169) | SSC 97 % | 99 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (con LOH de 1p y 16q )(n = 31) | SSC 87 % | 94 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (con LOH de 1p y 16q) y sin compromiso ganglionar (n = 12) | 92 % | 92 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (con LOH de 1p o 16q) y sin compromiso ganglionar (n = 13) | 85 % | 92 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (con LOH de 1p o 16q) y sin compromiso ganglionar (n = 68) | 87 % | 97 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| HF (con LOH de 1p o 16q) y compromiso ganglionar (n = 48) | 74 % | 92 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| AF | SSR 88 % | 100 % (n = 8) | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominal y régimen DD-4A |
| AF (tratamiento preoperatorio) | SSR 71 % | 71 % (n = 7) | Tratamiento preoperatorio con régimen DD-4A seguido de nefrectomía, muestreo de ganglios linfáticos y RT abdominal |
| AD | SSC 46 % | 53 % (n = 16) | Tratamiento preoperatorio con régimen I seguido de nefrectomía, muestreo de ganglios linfáticos y RT abdominal |
| AD | SSC 82 % | 91 % (n = 23) | Nefrectomía inmediata con muestreo de ganglios linfáticos seguido de RT abdominal y régimen UH1 |
Radioterapia
El inicio temprano de la radioterapia es un componente fundamental de la terapia multimodal para pacientes con tumor de Wilms no metastásico. En una revisión de 1488 pacientes con tumor de Wilms que se sometieron a cirugía y radioterapia, un intervalo superior a 14 días entre la cirugía y la radioterapia se relacionó con un riesgo más alto de mortalidad (CRI, 2,13; P = 0,013). Esto subraya la importancia de iniciar la radioterapia dentro de los 14 días posteriores a la cirugía, que se especifica en los protocolos de tratamiento del tumor de Wilms.[263][Nivel de evidencia C1]
Pérdida de heterocigosidad de 1p o 16q
En 635 pacientes con tumor de Wilms de tipo HF en estadio III que participaron en los protocolos del COG AREN0532 o AREN03B2, se observó que la pérdida de heterocigosidad de 1p o 16q afectó la SSC, pero no en la SG. La combinación del estado negativo de los ganglios linfáticos (análisis histológico) y un resultado negativo para la pérdida de heterocigosidad (relacionada con el tumor primario) fue un factor pronóstico sólido de SSC y SG excelentes.[275]
- Los pacientes sometidos a muestreo ganglionar durante la cirugía presentaron mejor SSC (tasa de la SSC a 4 años, 90,3 %) en relación con los pacientes que no se sometieron a muestreo ganglionar (tasa de SSC a 4 años, 80,0 %; P = 0,0037). No se observaron diferencias en la SG según el muestreo ganglionar (tasa de SC a 4 años, 97,0 % para pacientes con muestreo vs. 94,9 % para pacientes sin muestreo; P = 0,078).
- Los pacientes con compromiso ganglionar en el momento de la nefrectomía presentaron una SSC inferior que quienes no tenían compromiso ganglionar (83,5 vs. 94,2 % a los 4 años; CRI, 2,78; P = 0,00017). En la SG se observó un efecto limítrofe significativo (tasas de SG a 4 años, 95,1 vs. 98,2 %; CRI, 2,50; P = 0,054).
- En el Cuadro 7 se resume un análisis de los pacientes con tumor de Wilms en estadio III según el estado ganglionar (positivo para compromiso o negativo para compromiso) y la pérdida de heterocigosidad de 1p o 16q (positivo para uno, negativo para ambos).
Cuadro 7. Análisis de pacientes con tumor de Wilms en estadio III según el estado ganglionar y de LOH 1p o 16q Estado ganglionar LOH 1p o 16q Tasa de SSC a 4 añosa CRIb - = negativo; + = positivo; SSC = supervivencia sin complicaciones; CRI = cociente de riesgos instantáneos; LOH = pérdida de heterocigosidad. a Se observó una diferencia significativa en la SSC entre los grupos, en comparación con los pacientes con resultados negativos para compromiso ganglionar y LOH única (orden logarítmicoP< 0,0001). b El valor de supervivencia general no alcanzó significación estadística entre los dos grupos. - - 96,2 % - (n = 212) - +1p o 16q 89,5 % 3,04 (n = 72) + +1p 73,9 % 6,33 (n = 37) + +16q 79,3 % + - 86,0 % 3,57 (n = 104)
Pérdida de heterocigosidad de 1p y 16q
Se aumentó el tratamiento de los pacientes con pérdida de heterocigosidad de 1p y 16q incluidos en el ensayo AREN0533 (NCT00379340). Los pacientes con tumor de Wilms en estadio III y estadio IV con pérdida de heterocigosidad recibieron tratamiento con el régimen M. La tasa de SSC a 4 años fue del 90,2 %, y la tasa de SG fue del 96,1 %, en comparación con una tasa de SSC a 4 años del 61,3 % (P = 0,001) y una tasa de SG a 4 años del 86,0 % (P = 0,087) en los pacientes del ensayo NWTS-5. En el estudio se observó una mejora de la supervivencia, pero no tenía potencia suficiente para detectar diferencias en la supervivencia.[262][Nivel de evidencia C2]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Tratamiento del tumor de Wilms en estadio IV
En el Cuadro 8, se ofrece una visión general de las opciones de tratamiento estándar y los datos de supervivencia de pacientes con tumor de Wilms en estadio IV, a partir de resultados publicados.
| Tipo histológico | SSR o SSC a 4 años | SG a 4 años | Tratamientob |
|---|---|---|---|
| RC = respuesta completa; AD = tipo histológico anaplásico difuso; SSC = supervivencia sin complicaciones; AF = tipo histológico anaplásico focal; HF = tipo histológico favorable; LOH = perdida de heterocigosidad; SG = supervivencia general; SSR = supervivencia sin recaída; RT = radioterapia. | |||
| a Fuente: Grundy et al.,[109]Dome et al.,[182]Dix et al.,[264]Dix et al.,[262]Daw et al.[265]y Benedetti et al.[276] | |||
| b Para las descripciones del régimen de quimioterapia, consultar elCuadro 2. | |||
| c La RT abdominal se planifica de acuerdo con el estadio local del tumor renal. | |||
| d La RT pulmonar se reserva para los pacientes con indicios de metástasis pulmonares en la radiografía o la tomografía computarizada de tórax. | |||
| e Para obtener más información, consultar el estudioAREN0533 (NCT00379340). | |||
| HF (con nódulos pulmonares aislados) | SSC 85 % | 96 % | Nefrectomía con muestreo de ganglios linfáticos, seguido de RT abdominal,c con RT pulmonar bilateral o sin esta,d y régimen DD-4A o régimen Me |
| HF (sin LOH de 1p y 16q) con nódulos pulmonares aislados con RC a DD-4A | SSC 80 % | 96 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominalc y régimen DD-4A |
| HF (sin LOH de 1p y 16q) con nódulos pulmonares aislados con respuesta incompleta a DD-4A | SSC 99 % | 95 % | Nefrectomía con muestreo de ganglios linfáticos, seguido de RT abdominalc y RT pulmonar bilaterald, y régimen M. |
| HF (LOH de 1p y 16q) con nódulos pulmonares aislados (n = 18) | 100 % | 100 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominalc y RT pulmonar bilaterald, y régimen M. |
| HF (con LOH de 1p o 16q) (n = 20) | SSC 95 % | 100 % | Nefrectomía con muestreo de ganglios linfáticos, RT abdominalc radiación dirigida a sitios de metástasis, y régimen M |
| HF con metástasis extrapulmonares, y con metástasis pulmonares o sin estas | SSC 76 % | 89 % | Nefrectomía con muestreo de ganglios linfáticos seguido de RT abdominalc, régimen M y control local de otros sitios de metástasis; si hay metástasis pulmonares, RT pulmonar bilaterald |
| AF | SSC 61 % | 72 % (n = 11) | Nefrectomía con muestreo de ganglios linfáticos, seguido de RT abdominal,c radiación dirigida a sitios de metástasis, RT pulmonar bilateral,d y régimen DD-4A |
| AD | SSC 33 % | 33 % (n = 10) | Nefrectomía inmediata con muestreo de ganglios linfáticos seguido de RT abdominal,c radiación dirigida a sitios de metástasis, RT dirigida al pulmón completo,d y régimen I |
| AD (tratamiento preoperatorio) | SSC 60 % | 70 % (n = 10) | Tratamiento preoperatorio con régimen UH2 seguido de nefrectomía con muestreo de ganglios linfáticos, seguido de RT abdominal,c radiación dirigida a sitios de metástasis, y RT dirigida a todo el pulmónd |
La enfermedad en estadio IV se define por la presencia de metástasis hematógenas pulmonares, hepáticas, óseas, encefálicas o de otros sitios; el pulmón es el sitio más frecuente. La presencia de metástasis hepáticas en el momento del diagnóstico no es un factor pronóstico adverso independiente en pacientes con tumor de Wilms en estadio IV.[237]
Ganancia del cromosoma 1q
En el ensayo AREN0533 (NCT00379340), el 30 % de los pacientes con enfermedad pulmonar en estadio IV presentaron ganancia de 1q. Estos pacientes tuvieron una tendencia a una SSC más precaria, con independencia de la respuesta pulmonar y de si recibieron el régimen DD-4A o M.[264] Los pacientes de este ensayo con enfermedad pulmonar sola en estadio IV que presentaron una respuesta rápida completa a DD-4A y que no recibieron radiación pulmonar tuvieron una SSC más baja en comparación con los pacientes sin ganancia de 1q (tasas de SSC a 4 años, 57 vs. 86 %; P = 0,0013). La tasa de SG a 4 años, aunque no fue estadísticamente significativa, fue mejor en los pacientes sin ganancia de 1q en comparación con los pacientes con ganancia de 1q y enfermedad pulmonar (97 vs. 89 %; P = 0,16).[264] Los pacientes con respuesta incompleta lenta con enfermedad pulmonar sola y ganancia de 1q (tratados con DD-4A seguido de radioterapia pulmonar y régimen M) tuvieron tasas de SSC y SG a 4 años del 86 % y el 93 %, respectivamente. En comparación, los pacientes con respuesta incompleta lenta sin ganancia de 1q tuvieron tasas de SSC y SG a 4 años del 92 % y del 96 %, respectivamente.[264]
Tratamiento de los nódulos y las metástasis pulmonares
Tradicionalmente, se usaban radiografías de tórax para detectar las metástasis pulmonares. La introducción de la TC generó polémica porque en muchos pacientes se observaron nódulos pulmonares detectados mediante TC que no se podían observar en las radiografías de tórax. El abordaje de los pacientes con diagnóstico reciente de tumor de Wilms de tipo HF a quienes se les detectaron nódulos pulmonares solo mediante TC (radiografías de tórax negativas) ha sido motivo de controversia sobre la necesidad de un tratamiento intensivo adicional que se acompaña de efectos tóxicos agudos y crónicos.
Evidencia (tratamiento de nódulos pulmonares detectados solo en TC del tórax):
- En una revisión retrospectiva de 186 pacientes del NWTS-4 y el NWTS-5 con nódulos pulmonares detectados con TC sola, se notificó el uso de doxorrubicina, vincristina y dactinomicina versus el uso de 2 fármacos.[277]
- Los pacientes que recibieron doxorrubicina, vincristina y dactinomicina, con irradiación pulmonar o sin esta tuvieron una SSC a 5 años del 80 % versus el 56 % en los pacientes que recibieron dos fármacos (P = 0,004).
- No hubo diferencia en la SSC en función de la irradiación pulmonar.
- No hubo diferencia en la tasa de SG a 5 años (87 vs. 86 %).
En estudios retrospectivos europeos se examinó el efecto de omitir la radiación pulmonar en pacientes con metástasis pulmonares diagnosticadas por radiografía de tórax. Los investigadores europeos omitieron la radiación del tratamiento de la mayoría de los pacientes con tumor de Wilms y metástasis pulmonares identificadas mediante radiografía de tórax que se trataron en el ensayo SIOP-93-01 (NCT00003804). El abordaje europeo de los tumores renales se diferencia del abordaje que se utiliza en América del Norte. Todos los pacientes que exhiben un tumor renal en las pruebas con imágenes recibieron 9 semanas de quimioterapia con vincristina, dactinomicina y doxorrubicina antes de la nefrectomía.
Evidencia (omisión de la irradiación pulmonar):
- En un estudio retrospectivo de la SIOP, 234 pacientes con diagnóstico reciente de tumor de Wilms que presentaban metástasis pulmonares recibieron un tratamiento adaptado a la respuesta de las metástasis a la quimioterapia previa a la nefrectomía.[278]
- Los pacientes que estaban en remisión completa (67 %) al cabo de 6 semanas de tratamiento continuaron con la misma quimioterapia y no necesitaron radiación dirigida a los pulmones.
- La tasa de SSC a 5 años fue del 77 %, y la tasa de SG fue del 88 %.
- En los pacientes con metástasis pulmonares residuales se evaluó la necesidad de metastasectomía.
- Un grupo de 37 pacientes (17 %) alcanzó una remisión completa con la cirugía y su desenlace fue similar al del grupo de pacientes que recibieron quimioterapia. La viabilidad tumoral en las metástasis pulmonares resecadas no fue un factor para omitir la radioterapia.
- La tasa de SSC a 5 años fue del 84 %, y la tasa de SG fue del 92 %.
- Los pacientes con metástasis pulmonares residuales que se resecaron de forma incompleta o que eran inoperables recibieron quimioterapia más intensiva con ifosfamida o antraciclina alternada con carboplatino o etopósido durante 9 semanas.
- Los pacientes que exhibieron una remisión completa en ese momento no recibieron radiación pulmonar y continuaron con la quimioterapia, mientras que los pacientes con metástasis pulmonares residuales recibieron más quimioterapia (hasta completar 34 semanas) acompañada de irradiación pulmonar. La tasa de SG a 5 años fue del 48 % en comparación con las tasas de SG de los pacientes que respondieron a la quimioterapia sola (88 %) y los que se sometieron metastasectomía (92 %) (P <0,001).
- Los pacientes con tipos histológicos de riesgo alto, como el tumor de Wilms anaplásico, se trataron con quimioterapia más intensiva, pero tuvieron un resultado más deficiente en comparación con los pacientes con un tipo histológico no anaplásico (tasa de SG a 5 años, 33 vs. 87 %; P <0,001).
- Los pacientes que estaban en remisión completa (67 %) al cabo de 6 semanas de tratamiento continuaron con la misma quimioterapia y no necesitaron radiación dirigida a los pulmones.
- A partir de los resultados europeos, en el estudio del COG AREN0533 (NCT00379340) se usó una estrategia nueva para los pacientes con tumor de Wilms de tipo HF y metástasis pulmonares aisladas a fin de mejorar la SSC y reducir la exposición a la irradiación pulmonar. El tratamiento se ajustó de acuerdo con la respuesta de los nódulos pulmonares y la pérdida de heterocigosidad de 1p y 16q específica del tumor.[264][Nivel de evidencia C2]
- De los 292 pacientes inscritos en el estudio, 133 pacientes (42 %) exhibieron una respuesta completa del nódulo pulmonar después de 6 semanas de DD-4A (vincristina, dactinomicina y doxorrubicina) y continuaron recibiendo la misma quimioterapia sin radioterapia pulmonar. La tasa de SSC a 4 años fue del 80 %, y la tasa de SG fue del 96 %.
- Los pacientes con una respuesta incompleta de los nódulos pulmonares (n = 145) o pérdida de heterocigosidad de 1p/16q (n = 18) recibieron radioterapia pulmonar y 4 ciclos de ciclofosfamida y etopósido además de los fármacos del régimen DD-4A (régimen M). La tasa de SSC a 4 años fue del 89 % y la tasa de SG fue del 95 % en el grupo con respuesta incompleta de los nódulos pulmonares sin pérdida de heterocigosidad. En los pacientes con metástasis pulmonares aisladas y pérdida de heterocigosidad, la tasas de SSC a 4 años y SG fueron del 100 %.
- En un análisis posterior sobre la ganancia de 1q en 212 pacientes inscritos en AREN0533 de los que se disponía de DNA, los pacientes con remisión completa de los nódulos pulmonares con ganancia de 1q tuvieron una tasa de SSC a 4 años significativamente más bajas (86 vs. 57 %, P = 0,001) y una tendencia a tasas de SG inferiores (97 vs. 97 %). Hubo un predominio de recaídas pulmonares. No hubo diferencia en el desenlace de los pacientes con respuesta nodular pulmonar incompleta según la ganancia en 1q.
- El régimen M tiene un potencial más alto de efectos tardíos (aumento de riesgo de leucemias secundarias y riesgo de esterilidad relacionados con una dosis acumulada de ciclofosfamida de 8,8 g/m2).
- En el COG, se observó que la radioterapia pulmonar inicial se podría evitar en cerca del 40 % de los pacientes. La SG fue excelente; sin embargo, hubo una tendencia a más complicaciones que lo previsto (previsto del 15 % y observado del 20 %; P = 0,052).
Aunque se evitó la administración de radiación pulmonar a menos pacientes del ensayo del COG que de los ensayos europeos, es importante tener en cuenta varias diferencias entre los estudios y por este motivo no se pueden comparar de manera directa.[264,278] Los pacientes en Europa reciben un régimen de dosis más densas de dactinomicina y doxorrubicina, antes de la revaluación de las metástasis pulmonares, que los pacientes en América del Norte (135 ug/kg de dactinomicina y 100 mg/m2 de doxorrubicina en Europa, en comparación con 45 ug/kg de dactinomicina y 45 mg/m2 de doxorrubicina en América del Norte). En los estudios europeos se permite que se omita la radioterapia pulmonar en pacientes con una remisión completa después de la quimioterapia o metastasectomía pulmonar, mientras que la radioterapia solo se omite en los Estados Unidos para pacientes con remisión completa después de quimioterapia sola. Los estudios con imágenes no se revisaron de manera centralizada en los estudios europeos, pero sí en los de los Estados Unidos, y es posible que la definición de remisión completa fuera más estricta en el ensayo AREN0533 (NCT00379340).
Metástasis hepáticas en el momento del diagnóstico
El hígado es un sitio infrecuente de metástasis en el momento del diagnóstico de pacientes con tumor de Wilms de tipo HF en estadio IV, pero es el sitio más común después del pulmón. De 634 pacientes con tumor de Wilms de tipo HF en estadio IV de los estudios NWTS-4 y -5, 96 (15 %) presentaron compromiso hepático.[237] En el estudio AREN0533 de 47 pacientes (14 %) con tumor de Wilms de tipo HF que presentaban metástasis extrapulmonares, hubo 37 pacientes con metástasis hepáticas aisladas y 10 pacientes con metástasis hepáticas en combinación con otros sitios metastásicos. De los pacientes, 38 presentaron metástasis pulmonares y extrapulmonares. Todos los pacientes recibieron tratamiento con el régimen M y radioterapia dirigida al abdomen según el estadio tumoral local. La tasa de SSC a 4 años fue del 76 % y la de SG, del 89 %. Solo 2 pacientes se sometieron a resección de las metástasis hepáticas (ambos recibieron también radioterapia dirigida al hígado). De los pacientes que recibieron radioterapia dirigida al hígado (27 de 39), ninguno tuvo recidiva en dicho órgano.[276]
El efecto de las metástasis hepáticas en el momento del diagnóstico sobre el abordaje de la atención al paciente no se ha estudiado de manera prospectiva. La mayoría de las experiencias notificadas provienen de instituciones individuales o estudios retrospectivos que no incluyen abordajes quirúrgicos más modernos ni parámetros contemporáneos de estratificación del riesgo. En conjunto, no hay datos suficientes para apoyar que las metástasis en el hígado sean un sitio desfavorable de enfermedad metastásica, aunque hubo algunos estudios iniciales del SIOP que contradijeron este hallazgo.[237,279,280,281,282,283]
NWTS
En un análisis retrospectivo de 742 pacientes con tumor de Wilms en estadio IV que recibieron tratamiento en los ensayos NWTS-4 y 5, se examinaron los desenlaces de pacientes con metástasis pulmonares solas y sin estas. En el estudio también se investigaron los desenlaces de supervivencia de estos pacientes según si se sometieron a una resección de los tumores hepáticos.[237]
- De los pacientes, 22 se sometieron a resecciones hepáticas primarias (18 resecciones en cuña y 4 lobectomías). Después de la quimioterapia o la radioterapia, 13 pacientes se sometieron a resecciones hepáticas (7 resecciones en cuña, 5 lobectomías y 1 trisegmentectomía).
- Setenta y un pacientes (67 %) con enfermedad en el hígado no se sometieron a cirugía. En 14 pacientes, la enfermedad en el hígado respondió por completo a la quimioterapia sola.
- De los pacientes, 82 recibieron radioterapia dirigida al abdomen.
- La tasa de SSC fue del 75 % en los pacientes con tumor de Wilms de tipo HF metastásico. En los pacientes con metástasis solo pulmonares (n = 513), la tasa de SSC fue del 76 %. En los pacientes con metástasis hepáticas sin metástasis pulmonares (n = 34), la tasa de SSC fue del 76 %. La tasa de SSC fue del 70 % (n = 62) en los pacientes con metástasis hepáticas y pulmonares, en comparación con el 64 % en los pacientes (n = 25) con otros sitios de metástasis. No hubo diferencias significativas entre los grupos de pacientes en estadio IV (P = 0,60).
- La tasa de SSC fue del 86 % en los pacientes sometidos a resección primaria de metástasis hepáticas (n = 22), en comparación con el 68 % en los pacientes que no se sometieron a resección primaria de las metástasis hepáticas (P = 0,09).
- No se observaron diferencias significativas en la SSC en los pacientes tratados con quimioterapia sola en comparación con aquellos tratados con quimioterapia y radioterapia (P = 0,63). Las tasas de SSC fueron del 64 % en los pacientes que no recibieron radioterapia dirigida al abdomen, del 77 % en los pacientes que recibieron radioterapia dirigida al abdomen sin refuerzo y del 72 % en los pacientes que recibieron radioterapia dirigida al abdomen con refuerzo (P = 0,05).
- Los pacientes adultos formaron parte de la población del estudio (menos del 10 % eran mayores de 12 años y el 3 % eran mayores de 16 años).
- A partir de estos datos, los autores concluyeron que la presencia de metástasis hepáticas en el momento del diagnóstico no es un factor pronóstico adverso independiente para los pacientes con enfermedad en estadio IV. También indicaron que no hay razón para realizar una resección hepática en el entorno inicial. Estos 2 estudios se hicieron entre la décadas de 1960 y 1990. Durante este tiempo, las técnicas de resección hepática y los abordajes cambiaron a partir de una mejor comprensión de las características anatómicas del hígado. Además, los abordajes contemporáneos de estratificación del riesgo que tienen en cuenta las características citogenéticas del tumor no se evaluaron en estos estudios, ni tampoco las técnicas de radiación más convencionales, como la RTIM, que es posible que proporcione una mejor protección a los tejidos normales.[284]
SIOP
El grupo SIOP/Gesellschaft für Pädiatrische Hämatologie und Onkologie (GPOH) recomendó un abordaje quirúrgico más radical. Esta recomendación se basó en los desenlaces notificados de 29 pacientes tratados en los ensayos SIOP93-01/GPOH y SIOP2001/GPOH en los que se incluyeron 1365 pacientes entre 1994 y 2004. De los pacientes, 2 presentaron anaplasia difusa.[280]
- La tasa de SG a 5 años de los pacientes con metástasis hepáticas fue del 62,6 %, mientras que la tasa de SG de todos los pacientes con enfermedad en estadio IV fue del 76,3 %. Del grupo de pacientes con metástasis en el hígado, 14 no se sometieron a resecciones hepáticas. De estos pacientes, 4 (13 %) respondieron a la quimioterapia y seguían vivos en el momento de la publicación.
- Se sometieron a cirugía hepática 15 pacientes (11 resecciones hepáticas primarias, 6 resecciones completas y 5 resecciones incompletas). Sobrevivieron los 6 pacientes que se sometieron a una resección completa.
- Como resultado de estos hallazgos, los autores recomendaron un abordaje quirúrgico inicial radical para los pacientes con enfermedad hepática. Sin embargo, es importante recordar que los pacientes de SIOP reciben quimioterapia preoperatoria antes de la nefrectomía o la resección de lesiones metastásicas.
La cohorte francesa inscrita en el ensayo SIOP2001 incluyó a 131 pacientes con tumor de Wilms de tipo HF en estadio IV. De estos pacientes, 18 (14 %) presentaron metástasis hepáticas en el momento del diagnóstico, incluso 4 (3 %) con metástasis hepáticas aisladas.[283]
- La tasa de SSC a 5 años fue del 83 % y la tasa de SG fue del 88 % en esta cohorte de pacientes con metástasis hepáticas.
- Los autores concluyeron que el compromiso hepático no parece ser un marcador pronóstico adverso en pacientes con tumores de Wilms metastásicos, aunque en este estudio el número de pacientes fue pequeño.
Tratamiento del tumor de Wilms anaplásico difuso en estadio IV
En el estudio AREN0321 (NCT00335556), se probó la combinación de vincristina e irinotecán (VI) en un periodo inicial para pacientes con tumor de Wilms anaplásico difuso y enfermedad cuantificable.[265][Nivel de evidencia C2]
- Un grupo de 14 pacientes con tumor de Wilms anaplásico difuso en estadio IV recibieron la terapia del periodo inicial: 1 paciente logró una respuesta completa (RC), 10 pacientes lograron una respuesta parcial (RP) y ningún paciente logró una estabilización de la enfermedad. Lo anterior se tradujo en una tasa de RC y RP del 79 %.
- Los pacientes que respondieron a la combinación VI durante la terapia del periodo inicial incorporaron la combinación VI en el régimen terapéutico (UH2).
- Debido a la toxicidad cardíaca y pulmonar observada en este ensayo, el estudio se interrumpió y se modificó con dosis reducidas de doxorrubicina, ciclofosfamida y etopósido (cuando se combinó con carboplatino).
- Se planea otro estudio del régimen modificado en pacientes con tumor de Wilms anaplásico difuso recién diagnosticado.
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
- AREN1921 (NCT04322318) (A Study of Combination Quimiation for Patients with Diffuse anaplastic Tumors and Relapsed Favorable-Histology Wilms Tumors): este ensayo clínico tiene como objetivo mejorar la supervivencia al intensificar el tratamiento de los pacientes con tumores de Wilms anaplásicos difusos. En este ensayo se analizará más la mejora general observada con el régimen revisado UH1/UH2 del ensayo AREN0321, que se modificó junto con la adición de vincristina e irinotecán.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Tratamiento del tumor de Wilms en estadio V y de los pacientes con predisposición al tumor de Wilms bilateral
En la actualidad, no hay un abordaje estándar para el tratamiento de pacientes con tumor de Wilms en estadio V (tumor de Wilms bilateral en el momento del diagnóstico) ni de aquellos con predisposición al tumor de Wilms bilateral. Sin embargo, por primera vez, se completó un estudio prospectivo del tratamiento de pacientes con tumor de Wilms bilateral que proporciona orientación para el abordaje.[175]
El tratamiento de un niño con tumor de Wilms bilateral es muy difícil. Los objetivos del tratamiento son erradicar todo el tumor y preservar la mayor cantidad posible de tejido renal normal, con la esperanza de disminuir el riesgo de insuficiencia renal crónica en estos niños.[285]
Tradicionalmente, a partir de los ensayos NWTS-4 y NWTS-5, además de los ensayos europeos, los pacientes con tumor de Wilms bilateral exhiben una tasa de SSC y SG más bajas que los pacientes con tumor de Wilms localizado. En el estudio NWTS-4, se notificó que la tasa de SSC a 8 años en los pacientes con tumor de Wilms bilateral de tipo HF fue del 74 % y la tasa de SG fue del 89 %; en los pacientes con tipo histológico anaplásico, la tasa de SSC fue del 40 % y la tasa de SG fue del 45 %.[202] En el estudio NWTS-5, se notificó que la tasa de SSC a 4 años de todos los pacientes con tumor de Wilms bilateral fue del 56 % y la tasa de SG fue del 81 %; también se notificaron las tasas de SSC a 4 años de los pacientes con tipo HF (65 %), tipo histológico anaplásico focal (76 %) y tipo histológico anaplásico difuso (25 %).[109,182] En Europa se notificaron desenlaces similares para los pacientes con tumor de Wilms bilateral.[201,286] En una experiencia de una sola institución en los Países Bajos (N = 41), se observó una morbilidad significativa en términos de insuficiencia renal (32 %) y tumores secundarios (20 %).[286] La incidencia de insuficiencia renal en estadio terminal en el estudio realizado en los Países Bajos quizás sea reflejo de un período de seguimiento más largo.
Las opciones de tratamiento para el tumor de Wilms en estadio V (bilateral) son las siguientes:
Quimioterapia preoperatoria y resección
El objetivo del tratamiento para los pacientes con tumor de Wilms bilateral es conservar la mayor cantidad posible de tejido renal sin comprometer el desenlace general. Este abordaje se utiliza para evitar el efecto tardío de la enfermedad renal en estadio terminal, que puede obedecer a anomalías genéticas en la línea germinal subyacentes y a la pérdida de tejido renal funcional relacionada con el tratamiento. La enfermedad renal en estadio terminal se presenta con más frecuencia en pacientes con tumor de Wilms bilateral (12 % no sindrómico) que en pacientes con tumor de Wilms unilateral (<1 %). El desenlace renal funcional es bastante mejor después de la cirugía bilateral conservadora de nefronas que después de otros tipos de cirugía.[175]
Tradicionalmente, los pacientes se sometían a biopsias renales bilaterales, con estadificación de cada riñón, seguida de quimioterapia preoperatoria. En el primer ensayo prospectivo multicéntrico de tratamiento (COG AREN0534 [NCT00945009]), no se requirieron biopsias previas al tratamiento cuando los resultados de las pruebas de imágenes eran compatibles con tumor de Wilms.[175] Se decidió este abordaje porque la incidencia bilateral de tumores renales que no son de Wilms es muy baja. Además, las biopsias con aguja gruesa y las biopsias en cuña no son muy útiles para identificar la anaplasia en el tumor de Wilms.[173] En el entorno de una situación clínica inusual (como una edad mayor de 10 años o características atípicas en las imágenes), se debe considerar un diagnóstico distinto al tumor de Wilms, y se obtiene el diagnóstico mediante el análisis tisular.[175]
En los pacientes que reciben quimioterapia preoperatoria, es necesario evaluar las características patológicas del tumor después de 4 a 8 semanas. En los pacientes que no participan en ensayos clínicos, no está claro el momento ideal para hacer una biopsia o resección, porque una reducción mínima de tamaño a veces es consecuencia de una diferenciación inducida por la quimioterapia o de un tipo histológico anaplásico. Se lleva a cabo un intento planificado de resección o biopsia de un tumor aparentemente irresecable a más tardar 12 semanas después del diagnóstico. Cuando se continúa el tratamiento sin evaluar las características patológicas tumorales en un paciente con tumor de Wilms bilateral a veces se pasa por alto la presencia de anaplasia o diferenciación inducida por la quimioterapia (incluso diferenciación rabdomiomatosa) lo que conlleva aumento de la toxicidad sin proporcionar un beneficio adicional para el control tumoral. Las características histológicas anaplásicas se presentan en el 10 % de los pacientes con tumor de Wilms bilateral y estos tumores responden de manera precaria a la quimioterapia.[202]
Una vez se confirma el diagnóstico, se realiza una resección completa. La confirmación histológica del diagnóstico no es sencilla. En una serie de 27 pacientes del estudio NWTS-4, se observaron características patológicas discordantes (tumor anaplásico unilateral) en 20 casos (74 %), lo que destaca la necesidad de obtener tejido de ambos riñones. En 7 niños se obtuvieron biopsias con aguja gruesa con el fin de establecer el diagnóstico, y aunque no se encontró anaplasia, más tarde se confirmó el diagnóstico de tumores anaplásicos difusos. La anaplasia se identificó en sólo 3 de 9 pacientes cuando se realizó una biopsia abierta en cuña, y en 7 de 9 pacientes sometidos a una nefrectomía parcial o total.[202]
La decisión de administrar quimioterapia o radioterapia después de una biopsia o una revisión quirúrgica depende de la respuesta del tumor al tratamiento inicial. Se necesita un tratamiento más intensivo en pacientes con una respuesta inadecuada al tratamiento inicial, que se observa durante el segundo procedimiento o en el entorno de anaplasia.[214,287,288]
La enfermedad renal en estadio terminal es la complicación clínica más significativa en los pacientes con tumor de Wilms bilateral que a veces se debe a anormalidades en la línea germinal subyacentes o a una pérdida del tejido renal funcional debida al tratamiento. Es necesario realizar un control a largo plazo del funcionamiento renal después del tratamiento de una enfermedad bilateral.
Evidencia (quimioterapia preoperatoria y resección para el tumor de Wilms bilateral):
- En el primer estudio prospectivo sobre el tumor de Wilms bilateral (AREN0534 [NCT00945009]), se intentó mejorar la SSC y la SG al tiempo que se intentó preservar el tejido renal mediante la intensificación de la quimioterapia preoperatoria (con tres fármacos: Vincristina, dactinomicina y doxorrubicina), la cirugía definitiva a las 12 semanas del diagnóstico y la modificación de la quimioterapia posoperatoria en función de la respuesta histológica.[175]; [289][Nivel de evidencia C2]
- En el grupo de niños con tumor de Wilms bilateral, se observó que la revisión central de las imágenes, la resección quirúrgica dentro de las 12 semanas del diagnóstico y la terapia posoperatoria según la respuesta y las características histológicas, mejoraron la SSC y la SG en comparación con los desenlaces tradicionales en niños con tumor de Wilms bilateral.
- En los 180 pacientes con tumor de Wilms bilateral, la tasa de SSC a 4 años fue del 81 % (intervalo de confianza [IC] 95 %, 74 – 87%) y la tasa de SG fue del 95 % (IC 95 %, 91 – 99%). El abordaje descrito antes con asignación de riesgo para el tratamiento a partir de la estadificación y la histopatología posoperatoria produjo resultados excelentes en los pacientes con tumores de Wilms bilaterales, pero no mejoró el desenlace de los pacientes con anaplasia difusa. La tasa de SSC a 4 años de 7 pacientes con tumores completamente necróticos fue del 100 %. En 118 pacientes con tumores histopatológicos de riesgo intermedio, la tasa de SSC a 4 años fue del 82 % y la tasa de SG fue del 97 %. Catorce pacientes con tumores de tipo blastémico presentaron una tasa de SSC a 4 años del 79 % y una tasa de SG del 93 %. Los pacientes con necrosis completa se asignaron a la categoría de riesgo bajo y aquellos con características histopatológicas de tipo blastémico a la categoría de riesgo alto para los tratamientos posteriores. En 18 pacientes con anaplasia difusa, la tasa de SSC a 4 años fue del 61 % y la tasa de SG fue del 72 %. En 7 pacientes con anaplasia focal, la tasa de SSC fue del 71 % y la tasa de SG fue del 100 %.[289][Nivel de evidencia C2] Debido a que no se realizó una biopsia antes del tratamiento en esta serie, es posible que algunos de los pacientes inscritos solo tuvieran restos nefrogénicos y no un tumor de Wilms verdadero. Puede que este hallazgo haya mejorado las cifras de supervivencia en comparación con los controles históricos.
- Uno de los objetivos del estudio fue que el 75 % de los pacientes se sometieran a la cirugía definitiva dentro de 12 semanas. Después de la quimioterapia de inducción, 163 de 189 pacientes (84 %) se sometieron al tratamiento quirúrgico definitivo por lo menos en un riñón a las 12 semanas, y el 39 % de los pacientes conservaron partes de ambos riñones.
- La quimioterapia posterior a la cirugía se diseñó de acuerdo con la respuesta histológica. La tasa de SSC a 4 años fue de 84,1 % para los tumores de tipo HF, de 58,2 % para los tumores de tipo histológico anaplásico y de 82 % para los tumores de tipo blastémico.
- Debido al riesgo más alto de insuficiencia renal en los pacientes con tumor de Wilms bilateral en comparación con el tumor de Wilms unilateral, uno de los objetivos del estudio fue que 50 % de los pacientes se sometieran a cirugía bilateral conservadora de nefronas. Este umbral no se alcanzó, y solo 39 % de los pacientes se trataron con éxito con cirugía bilateral conservadora de nefronas.
- A partir del estudio anterior, la recomendación fue continuar la quimioterapia preoperatoria de 3 fármacos durante 6 a 12 semanas, y luego cirugía conservadora de nefronas, siempre que fuera posible. Después de la resección, la terapia posoperatoria se basó en las características histológicas de la pieza extirpada. El resultado decepcionante de la cirugía conservadora de nefronas en este estudio quizás se explique por el grado de experiencia de los cirujanos en este estudio multicéntrico.
- En el estudio AREN0534 se examinaron los desenlaces de pacientes con anaplasia y tumor de Wilms bilateral. Se incluyeron 27 pacientes (17 con anaplasia difusa y 10 con anaplasia focal). De los pacientes, 26 presentaron enfermedad bilateral y 1 paciente presentó enfermedad unilateral (en el único riñón del paciente).[290]
- De los 26 pacientes con enfermedad bilateral, 21 presentaron características patológicas discordantes. Esto indica que, si es necesaria una biopsia, deben tomarse muestras de ambos riñones.
- Las tasas de SSC a 4 y 8 años fueron del 53 % para los pacientes con anaplasia difusa. La tasa de SSC a 4 años fue del 80 % y la tasa de SSC a 8 años fue del 70 % para los pacientes con anaplasia focal.
- La SSC y la SG no difirieron según el estado de los márgenes.
- Todos los niños que murieron experimentaron una recaída o progresión previa en los 18 meses anteriores a la inscripción en el estudio.
- En una revisión retrospectiva de 49 pacientes con tumor de Wilms que recibieron terapia preoperatoria de acuerdo con las directrices de SIOP-93-01 (NCT00003804), se determinó que el momento oportuno para la cirugía era cuando ya no se observaban indicios de regresión tumoral en las imágenes. La media de duración del tratamiento fue de 80 días antes de la cirugía con conservación del riñón.[291]
- La tasa de SSC a 5 años fue del 83,4 %, y la tasa de SG fue del 89,5 %.
- Todos los pacientes, excepto uno, se sometieron a cirugía con conservación de por lo menos un riñón.
- Pese a la buena tasa de supervivencia, el 14 % de los pacientes presentaron enfermedad renal en estadio terminal.
- En una revisión retrospectiva del St. Jude Children's Research Hospital, los investigadores describieron su experiencia con la quimioterapia preoperatoria seguida de procedimientos para conservar el riñón en niños con tumor de Wilms bilateral de tipo HF.[292]
- En una serie, 39 de 42 pacientes con tumor de Wilms bilateral de tipo HF se sometieron con éxito a procedimientos bilaterales de conservación de los riñones después de recibir quimioterapia preoperatoria. En 3 pacientes, se realizó una nefrectomía unilateral con cirugía conservadora de nefronas contralateral. Fue necesario repetir la cirugía conservadora de nefronas en 3 pacientes de manera anticipada (dentro de los primeros 4 meses) por un tumor residual. A largo plazo, 7 pacientes presentaron recidiva local del tumor y 3 pacientes sufrieron obstrucción intestinal.
- La tasa de SG fue del 86 % (media de seguimiento, 4,1 años). De los 6 pacientes que murieron, 5 exhibían un tipo histológico anaplásico difuso.
- Todos los pacientes tuvieron una tasa de filtración glomerular estimada de más de 60 ml/min/1,73m2 en el último seguimiento; ninguno de los pacientes presentó enfermedad renal en estadio terminal.
- Los autores concluyeron que la cirugía bilateral para conservar el riñón casi siempre es factible, y que se puede realizar de forma inocua con buenos desenlaces oncológicos en pacientes con tumor de Wilms bilateral sincrónico. Esta cirugía se deberá considerar aunque los estudios con imágenes preoperatorias indiquen que las lesiones son irresecables. Es posible que la conservación del parénquima renal ayude a mantener el funcionamiento renal de los niños que tienen un riesgo importante de insuficiencia renal crónica. Es necesario un seguimiento cuidadoso a largo plazo para evaluar la posibilidad de progresión a disfunción renal.
- En una revisión de seguimiento de estos pacientes, se revelaron los siguientes aspectos: 8 de 36 pacientes debieron repetir la cirugía conservadora de nefronas y otros 2 pacientes requirieron de una tercera cirugía de este tipo. Seis de estos pacientes seguían vivos y sin enfermedad en el seguimiento a 4,5 años. Los 2 pacientes que fallecieron tenían un tipo histológico de predominio blastémico.[293]
Para obtener información sobre la enfermedad recidivante, consultar la sección Tratamiento y desenlaces del tumor de Wilms recidivante.
Tratamiento de pacientes con tumores de Wilms multicéntricos o tumores unilaterales con predisposición a tumores bilaterales
A partir de una subpoblación de pacientes con tumor de Wilms que tienen riesgo de enfermedad metacrónica además de un riesgo más alto de enfermedad renal terminal, el COG dirigió el estudio prospectivo más extenso (AREN0534 [NCT00945009]) en estos pacientes. El objetivo de este estudio fue preservar el tejido renal y mantener excelentes desenlaces generales.[4,91]
Los pacientes se identificaron en la institución de tratamiento como pacientes con síndromes de predisposición. La quimioterapia de inducción se determinó por la presencia de enfermedad localizada o metastásica identificada en las imágenes (además del tipo histológico si se contaba con una biopsia) en el momento del diagnóstico. La cirugía, incluso la cirugía con conservación del riñón, se basó en la respuesta radiográfica a las 6 o 12 semanas, y la quimioterapia adicional se determinó a partir del tipo histológico. Los pacientes con enfermedad en estadio III o IV de tipo histológico favorable y todos los pacientes con anaplasia recibieron radioterapia.[146][Nivel de evidencia C1]
- Se inscribieron 34 pacientes con las siguientes afecciones subyacentes: síndrome de Beckwith-Wiedemann (n = 9), hemihipertrofia (n = 9), tumores multicéntricos (n = 10), síndrome WAGR (n = 2), riñón único (n = 2), síndrome de Denys-Drash (n = 1) y síndrome de Simpson-Golabi-Behmel (n = 1).
- Las tasas de SSC a 4 años y SG fueron del 94 % y del 100 %, respectivamente, con una mediana de seguimiento de 4,49 años. Se presentaron recaídas en 2 pacientes (una en el lecho tumoral y otra en el abdomen); ninguna de las muertes ocurrió durante la inducción.
- La quimioterapia antes de la nefrectomía facilitó la conservación renal en 22 de los 34 pacientes (65 %). Se realizaron 11 nefrectomías parciales después de 2 ciclos de quimioterapia, y 9 nefrectomías parciales después de 4 ciclos de quimioterapia. De los tumores, 2 se resolvieron por completo después de la quimioterapia y no requirieron de cirugía posterior.
- Un grupo de 22 pacientes tenía un síndrome de predisposición conocido para el que se habría previsto exámenes de detección con ecografía. De estos pacientes, 16 tenían enfermedad en estadio I, 3 tenían enfermedad en estadio II y otros 3 tenían enfermedad en estadio III. De todos los tumores, 13 se detectaron durante una ecografía de rutina.
- Estos resultados indican que un abordaje de tratamiento estandarizado con quimioterapia preoperatoria, resección quirúrgica dentro de las 6 a las 12 semanas y quimioterapia posoperatoria según el tipo histológico se traduce en una SSC y SG excelentes, además de conservación del parénquima renal.
Trasplante renal
El trasplante renal en niños con tumor de Wilms en estadio V se suele posponer hasta que hayan pasado entre 1 y 2 años sin indicios de neoplasia maligna porque la mayoría de las recaídas ocurren dentro de los 2 años que siguen al diagnóstico.[294] De manera similar, el trasplante renal en niños con síndrome de Denys-Drash y tumor de Wilms, que requieren nefrectomía bilateral, por lo general se retrasa de 1 a 2 años después de completar el tratamiento inicial.[294]
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Tratamiento y desenlaces del tumor de Wilms recidivante
Para los pacientes con tumor de Wilms recidivante se debe considerar la participación en ensayos clínicos de fase I y fase II disponibles. A continuación se describen otras opciones de tratamiento para el tumor de Wilms recidivante.
Los cuidados paliativos siguen siendo un enfoque central del tratamiento, con independencia de si se sigue con la terapia dirigida a la enfermedad en el momento de la progresión. Esto garantiza que se aumenta al máximo la calidad de vida mientras se intenta reducir el estrés de la enfermedad terminal.
Pronóstico, factores pronósticos y categorías de riesgo del tumor de Wilms recidivante
Alrededor del 15 % de los pacientes con tumor de Wilms de tipo HF y el 50 % de los pacientes con tumor de Wilms de tipo histológico anaplásico presentan recidiva.[179] El sitio más común de recaída es el pulmón, seguido del abdomen o el flanco, y el hígado. La recidiva en el encéfalo (0,5 %) o en el hueso es poco frecuente en los niños con tumor de Wilms.[295,296] Tradicionalmente, la tasa de rescate de los pacientes con tumor de Wilms de tipo HF recidivante fue del 25 % al 40 %. Como resultado de las combinaciones modernas de tratamiento, el desenlace después de la recidiva ha mejorado hasta el 60 %.[297,298]
Cerca del 95 % de las primeras recidivas tumorales de Wilms se presentan dentro de los 2 años del diagnóstico inicial. La recaída que ocurre más de 5 años después del diagnóstico se considera una recidiva tardía y es poco frecuente. En el estudio retrospectivo más largo con más de 1300 niños inscritos en varios ensayos de tumor de Wilms, la mediana de tiempo hasta la recidiva tardía después de la primera recidiva fue de 13 años (intervalo, 5–17 años).[299,300]
Se analizó un número de características pronósticas que afectan los desenlaces después de una recidiva, pero es difícil determinar si estos factores son independientes entre sí. Además, los siguientes factores pronósticos cambian conforme evoluciona el tratamiento del tumor de Wilms primario y recidivante:
- Tipo histológico anaplásico.[301]
- Estadio avanzado del tumor.[301]
- Sexo. El sexo fue un factor predictivo del desenlace, la evolución de los hombres fue más desfavorable que la de las mujeres.[297,302]
En el ensayo NWTS-5 se observó que el tiempo hasta la recidiva y el sitio de recidiva ya no son significativos para el pronóstico.[297,302] Sin embargo, en un estudio SIOP, los pacientes que presentaron una recaída pulmonar dentro de los 12 meses del diagnóstico tuvieron un pronóstico más precario (tasa de SG a 5 años, 47 %) que los pacientes que presentaron una recaída pulmonar 12 meses o más después del diagnóstico (tasa de SG a 5 años, 75 %).[303]
A partir de estos resultados, se identificaron las siguientes 3 categorías de riesgo:
- Riesgo estándar: pacientes con tumor de Wilms de tipo HF que recaen luego de recibir tratamiento solo con vincristina o dactinomicina. Estos pacientes representan cerca del 30 % de las recidivas y se espera que tengan tasas de SSC del 70 % al 80 %.[298]
- Riesgo alto: pacientes con tumor de Wilms de tipo HF que recaen luego de un tratamiento con 3 o más fármacos. Estos pacientes representan del 45 % al 50 % de los niños con tumor de Wilms que recaen y tienen tasas de supervivencia que oscilan entre el 40 % y el 50 %.[298]
- Riesgo muy alto: pacientes con tumor de Wilms con predominio anaplásico o blastémico recidivante. Estos pacientes representan entre el 10 % y el 15 % de todas las recaídas de tumor de Wilms y se espera que tengan tasas de supervivencia cercanas al 10 %.[182,298]
Tratamiento del tumor de Wilms de riesgo estándar en recaída
En los niños que presentaron un tumor de Wilms pequeño en estadio I tratados con cirugía sola, la tasa de SSC fue del 84 %. Todos los niños que recayeron, excepto uno, se rescataron con un tratamiento adaptado al sitio de la recidiva.[183,302]
Es posible volver a administrar tratamiento con éxito a los pacientes en recaída que presentaron un tumor de Wilms tratado al inicio con nefrectomía inmediata, seguida de quimioterapia con vincristina y dactinomicina.
Las opciones de tratamiento del tumor de Wilms de riesgo estándar en recaída son las siguientes:
Cirugía, radioterapia y quimioterapia
Evidencia (cirugía, radioterapia y quimioterapia):
- En 58 pacientes se usó el protocolo de recaída del NWTS-5 con escisión quirúrgica cuando fue posible, además de radioterapia y cursos de vincristina, doxorrubicina y ciclofosfamida alternados con etopósido y ciclofosfamida.[302]
- La tasa de SSC a 4 años después de la recaída fue del 71 %, y la tasa de SG fue del 82 %.
- En los pacientes que solo presentaron una recidiva pulmonar, la tasa de SSC a 4 años fue del 68 % y la tasa de SG fue del 81 %.
- El SIOP Renal Tumor Study Group (SIOP-RTSG) analizó los desenlaces de los pacientes de riesgo bajo e intermedio (n = 109) que recayeron después del tratamiento con vincristina y dactinomicina para el tumor de Wilms primario en los estudios SIOP 93-01 y SIOP 2001.[304][Nivel de evidencia C1]
- La tasa de SSC a 5 años fue del 72,3 %, y la tasa de SG fue del 79,3 %.
- Los pacientes tratados con vincristina, dactinomicina y doxorrubicina (VAD) no tuvieron desenlaces más precarios que los pacientes tratados con terapias más intensivas (como ciclofosfamida, carboplatino, etopósido y doxorrubicina o ifosfamida, carboplatino y etopósido [ICE]; CRI para la SSC, 0,611; CRI para la SG, 0,438).
- No hubo diferencia de supervivencia entre los pacientes tratados con VAD en comparación con los regímenes de ICE.
- El tipo de alquilante (ifosfamida o ciclofosfamida) utilizado en regímenes de tratamiento más intensivos no afectó las tasas de supervivencia.
- La incorporación de dosis altas de trasplante de células madre hematopoyéticas (TCMH) para consolidar el tratamiento de la recaída no mejoró el desenlace de los pacientes con recaída de riesgo estándar.
- Es posible que en estudios futuros se identifiquen pacientes que se pueden beneficiar del tratamiento con VAD en lugar de un tratamiento más intensivo con quimioterapia combinada, como ciclofosfamida, carboplatino, etopósido y doxorrubicina.
Tratamiento del tumor de Wilms de riesgo alto y riesgo muy alto en recaída
Las opciones de tratamiento del tumor de Wilms de riesgo alto y de riesgo muy alto en recaída son las siguientes:
Quimioterapia, cirugía o radioterapia
Evidencia (quimioterapia, cirugía o radioterapia):
- Es posible volver a tratar con éxito a cerca del 50 % de los pacientes con tumor de Wilms unilateral que presentaron recaída o progresión luego del tratamiento inicial con vincristina, dactinomicina, doxorrubicina y radioterapia. Sesenta pacientes con tumor de Wilms unilateral se trataron en el protocolo de recaída NWTS-5 con ciclos alternos de ciclofosfamida con etopósido y carboplatino con etopósido, cirugía y radioterapia.[297][Nivel de evidencia B4]
- La tasa de SSC a 4 años de los pacientes con tumor de Wilms de riesgo alto fue del 42 % y la tasa de SG fue del 48 %.
- Los pacientes de riesgo alto que recayeron solo en los pulmones, tuvieron una tasa de SSC a 4 años del 49 % y una tasa de SG del 53 %.
Los pacientes con tumores anaplásicos en estadio II, estadio III y estadio IV en el momento del diagnóstico tienen un pronóstico muy adverso en el momento de la recidiva.[182] Se demostró actividad con la combinación de ifosfamida, etopósido y carboplatino en este grupo de pacientes, pero se observaron efectos tóxicos hematológicos significativos.[305]
Trasplante de células madre hematopoyéticas
Se usaron dosis altas de quimioterapia seguidas de trasplante de células madre hematopoyéticas (TCMH) autógeno en pacientes de riesgo alto que recidivan.[306,307]; [308,309][Nivel de evidencia C1]
Evidencia (trasplante de células madre hematopoyéticas):
- Los investigadores utilizaron el European Blood and Marrow Transplantation Registry para examinar los desenlaces de los niños con tumor de Wilms (n = 69) que recibieron dosis altas de quimioterapia con TCMH autógeno como terapia de consolidación durante la primera o la segunda remisión. Se usaron diferentes regímenes de quimioterapia de dosis altas que contenían melfalán (n = 34) o tiotepa (n = 14).[310][Nivel de evidencia C1]
- Las probabilidades de SG y SSC a 5 años fueron de 0,67 (± 0,06) y 0,63 (± 0,06), respectivamente (mediana de tiempo de observación, 7,8 años).
- Los niños que se sometieron a trasplante en la primera remisión tuvieron probabilidades de SG y SSC a 5 años de 0,69 y 0,72, respectivamente.
- El uso de melfalán solo para dosis altas de quimioterapia no condujo a tasas de supervivencia inferiores, en comparación con otros medicamentos o combinaciones de medicamentos, y produjo una mejor incorporación del injerto, en comparación con los regímenes que contenían tiotepa.
- Se notificaron desenlaces similares en una serie de 54 pacientes con tumor de Wilms que recibieron dosis altas de quimioterapia con rescate de células madre autógenas (terapia de primera línea, n = 13; terapia de recidiva, n = 41).[309][Nivel de evidencia C1]
- En los pacientes que se trataron al momento de la recidiva, las tasas de SSC y SG a 5 años fueron del 57 % y del 69 %, respectivamente (mediana de seguimiento, 7 años).
- El Center for International Blood and Marrow Transplantation Research recabó y analizó los desenlaces de 253 pacientes con tumor de Wilms en recaída que recibieron dosis altas de quimioterapia, seguidas de TCMH autógeno entre 1990 y 2013.[311]
- La tasa de SSC a 5 años fue del 36 % y la tasa de SG a 5 años fue del 45 %.
- La recaída de la enfermedad primaria fue la causa de muerte en el 81 % de la población.
No se han notificado ensayos aleatorizados en los que se compare la quimioterapia con el trasplante, y las series de casos presentan sesgo de selección.
A todos los pacientes en quienes fracasan los intentos de rescate se les debería ofrecer tratamiento en los ensayos clínicos de fase I o fase II disponibles.
Opciones de tratamiento en evaluación clínica para el tumor de Wilms recidivante
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
- AREN1921 (NCT04322318) (A Study of Combination Quimiation for Patients with Diffuse anaplastic Tumor and Relapsed Favorable-Histology [FH] Wilms Tumor): los pacientes con tumor de Wilms de tipo HF en recaída de riesgo estándar (tratados antes con 2 medicamentos) recibirán quimioterapia de vincristina, doxorrubicina y ciclofosfamida en alternancia con ciclofosfamida, carboplatino y etopósido alternados con vincristina e irinotecán Los pacientes con tumor de Wilms en recaída de riesgo alto (tratados antes con 3 medicamentos) y con tumor de Wilms de tipo HF en recaída de riesgo muy alto (tratados antes con 4 o más medicamentos) se tratarán con ciclofosfamida y topotecán añadidos a un tratamiento de base con ifosfamida, carboplatino y etopósido.
Seguimiento posterior al tratamiento
Los pacientes que completaron el tratamiento para el tumor de Wilms y que presentan características compatibles con una predisposición genética, como un tumor de Wilms bilateral, requieren exámenes de detección con ecografía renal cada 3 meses para identificar tumores metacrónicos durante el período de riesgo para el síndrome especifico (5 años para los síndromes relacionados con WT1; 8 años para el síndrome de Beckwith-Wiedemann).
Efectos tardíos posteriores al tratamiento del tumor de Wilms
Los niños tratados por tumor de Wilms tienen un aumento de riesgo de presentar los siguientes efectos:
- Mortalidad prematura después del diagnóstico de tumor de Wilms. En el Childhood Cancer Survivor Study (CCSS), se evaluó la morbilidad y mortalidad a largo plazo entre los sobrevivientes de tumores de Wilms unilaterales no sindrómicos. Entre 2008 sobrevivientes, se presentaron 142 muertes (razón estandarizada de mortalidad [REM], 2,9 [IC 95 %, 2,5–3,5]; incidencia acumulada de muerte a 35 años, 7,8 % [IC 95 %, 6,3–9,2 %]). Las causas más frecuentes de muerte fueron las neoplasias malignas subsiguientes (NMS) (n = 42), la recaída del tumor de Wilms (n = 30) y las relacionadas con problemas cardíacos (n = 9). Los sobrevivientes tratados solo con vincristina y dactinomicina (VA) tuvieron un riesgo de mortalidad por todas las causas (REM,1,0) y mortalidad tardía relacionada con la salud (REM, 1,5) comparables con los de la población general.[312]
- Afecciones crónicas. Los resultados del mismo estudio del CCSS son los siguientes:[312]
- Entre los sobrevivientes de tumor de Wilms unilateral (n = 2008), la incidencia acumulada a 35 años de cualquier afección crónica de grados 3 a 5 fue del 34,1 %, en comparación con el 14,8 % entre sus hermanos de ambos sexos.
- Los sobrevivientes tratados solo con VA tuvieron un riesgo ligeramente mayor de presentar afecciones crónicas de grado 3 a 5 en comparación con los hermanos de ambos sexos (RR, 1,5).
- Los riesgos de afecciones crónicas de grado 3 a 5, como obstrucción intestinal (8,1 %), insuficiencia renal (2,4 %), insuficiencia ovárica prematura (7,3 %) e insuficiencia cardíaca (4,0 %), aumentaron con la intensidad del tratamiento en función de la dosis y por la exposición a doxorrubicina o radioterapia. Este riesgo fue más pronunciado entre los sobrevivientes con enfermedad de riesgo alto, incluso en aquellos con recaída temprana (<5 años).
- En comparación con los hermanos, los sobrevivientes tratados con VA presentaron un riesgo más alto de obstrucción intestinal (RR, 9,4) e insuficiencia renal (RR, 11,9), pero la magnitud del riesgo fue más baja que la de los grupos de tratamiento más intensivo.
- Tanto la radioterapia dirigida a todo el pulmón como la dirigida a todo el abdomen se relacionaron con un aumento del riesgo de NMS, insuficiencia cardíaca y obstrucción intestinal, y la magnitud del riesgo aumentó con el aumento de las dosis.
- Las dosis de radioterapia dirigida a todo el abdomen o el flanco superiores a 20 Gy se relacionó con un aumento del riesgo de obstrucción intestinal en comparación con la ausencia de radioterapia, mientras que las dosis de radioterapia de 20 Gy o inferiores no se relacionaron con ninguna afección crónica.
- Neoplasias malignas subsiguientes.[313,314] En el estudio del CCSS de sobrevivientes de tumor de Wilms (n = 2008), 82 presentaron neoplasias malignas subsiguientes (NMS) (cociente de incidencia estandarizada [CIE], 4,1), lo que representa una tasa de incidencia acumulada del 6,1 %. Los cánceres mamarios (CIE, 6,9), tiroideos (CIE, 4,7) e intestinales o colorrectales (CIE, 12,0) fueron los que se notificaron con más frecuencia.[312] La incidencia acumulada de cáncer de mama invasivo en sobrevivientes de tumor de Wilms que recibieron radiación pulmonar por tumor de Wilms metastásico es de casi el 15 % a los 40 años de edad.[315]
- Insuficiencia cardíaca congestiva. El riesgo de insuficiencia cardíaca congestiva se modifica según la dosis de doxorrubicina recibida, la radiación dirigida al corazón y el sexo femenino.[316] Los investigadores del CCSS notificaron que las dosis acumuladas de doxorrubicina de 250 mg/m2 o superiores se relacionaron con una tasa casi 5 veces mayor de aparición de insuficiencia cardíaca, en comparación con la ausencia de exposición a la doxorrubicina.[312]
- Infertilidad en los pacientes que recibieron irradiación dirigida a todo el abdomen o ciclofosfamida.[317,318] La tasa de incidencia acumulada a 35 años de insuficiencia ovárica prematura fue del 7,3 %. La radioterapia dirigida a todo el abdomen se relacionó con un aumento del riesgo de insuficiencia ovárica prematura; el riesgo se incrementó al aumentar la dosis (≤20 Gy: RR, 13,1; >20 Gy: RR, 36,5).[312] Las mujeres sobrevivientes de tumor de Wilms diagnosticado antes de los 40 años de edad en el St Jude Lifetime Cohort Study experimentaron un aumento de la insuficiencia ovárica prematura en comparación con los controles (9,3 vs. 0,6 %; P < 0,01), probablemente debido a la irradiación abdominal que expuso los ovarios. Ninguna de las mujeres expuestas a radioterapia hemiabdominal presentó insuficiencia ovárica prematura.[314]
- Complicaciones del embarazo.[319] En un estudio se evaluaron los efectos de la radioterapia dirigida al abdomen (todo el abdomen o parte de este) en el embarazo y los desenlaces del embarazo en pacientes con antecedentes de tumor de Wilms. Los investigadores evaluaron la hipertensión que complica el embarazo, el parto prematuro o la amenaza de parto, la posición anómala del feto, la rotura prematura de membranas, el parto obstruido, la anormalidad de las fuerzas del parto y las complicaciones del cordón umbilical. Hubo 1021 embarazos (con una duración de 20 semanas o más), con 955 bebés nacidos vivos, de los que se obtuvieron 700 conjuntos de registros médicos de las madres y los descendientes. Las mujeres sobrevivientes de tumor de Wilms que recibieron radioterapia tienen un riesgo mayor de hipertensión que complica el embarazo, amenaza de parto o parto prematuro, y posición anómala del feto. Los descendientes de estos pacientes tenían más probabilidades de ser prematuros y tener bajo peso al nacer.[319] Para obtener más información, consultar la sección Reproducción en Efectos tardíos del tratamiento anticanceroso en la niñez.
- Insuficiencia renal tardía. De acuerdo al estudio del CCSS, la incidencia acumulada de insuficiencia renal tardía en los sobrevivientes fue del 2,4 % (una tasa cerca de 10 veces a la de los hermanos de ambos sexos). Ningún tipo de exposición a quimioterapia o a la radioterapia se relacionó con insuficiencia renal, lo que indica que la nefrectomía quizás sea el factor de riesgo principal.[312] Las tasas de insuficiencia renal tardía aumentan con la edad avanzada. Este hallazgo destaca la importancia del seguimiento a largo plazo de la función renal en estos pacientes, en especial, cuando tienen otras afecciones comórbidas. La incidencia acumulada de enfermedad renal en estadio terminal causada por insuficiencia renal crónica a los 20 años del diagnóstico del tumor de Wilms es del 3,1 % en los pacientes con tumor de Wilms bilateral.[211]
- Deterioro neurocognitivo. Entre 264 sobrevivientes a largo plazo de tumor de Wilms que participaron en un estudio de St Jude Lifetime Cohort Study y que completaron las pruebas neurocognitivas, el 19,7 % presentó deterioro de la función ejecutiva de grados 2 a 3, en comparación con el 12 % de los controles (P <0,01). En los sobrevivientes, la velocidad de procesamiento (19,7 vs. 8,4 %) y la memoria (20,9 vs. 9,9 %) mostraron una prevalencia más alta de alteraciones moderadas a graves, en comparación con los controles (P < 0,01). Solo las alteraciones en la velocidad de procesamiento se relacionaron con la presencia de enfermedad cardiovascular de grados 2 a 4.[314]
- Rendimiento físico. En el estudio del CCSS, los sobrevivientes tuvieron más probabilidades de notificar un funcionamiento físico deficiente (OR, 2,9) que los hermanos.[312] En un estudio de la St Jude Lifetime Cohort, 270 sobrevivientes a largo plazo de tumor de Wilms completaron las pruebas de funcionamiento y se observó que presentaban un exceso de alteraciones en la función aeróbica, la movilidad, la fuerza, la resistencia y la flexibilidad, en comparación con los controles. No hubo una relación clara entre estos desenlaces y las exposiciones del tratamiento.[314]
Los efectos tardíos renales en los pacientes con tumor de Wilms y anomalías genéticas subyacentes son los siguientes:
- Los niños con síndrome WAGR u otras variantes germinales de WT1 se vigilan durante toda la vida porque tienen un mayor riesgo de presentar hipertensión, nefropatía e insuficiencia renal.[211]
- Los pacientes con tumor de Wilms y aniridia sin anomalías genitourinarias tienen un riesgo más bajo, pero se deben vigilar por nefropatía o insuficiencia renal.[320]
- Los niños con tumor de Wilms y cualquier anomalía genitourinaria también tienen un mayor riesgo de insuficiencia renal tardía y se deben vigilar. Las características relacionadas con las variantes germinales de WT1 que aumentan el riesgo de presentar insuficiencia renal son las siguientes:[211]
- Tipo histológico de predominio estromal.
- Enfermedad bilateral.
- Restos nefrogénicos intralobulares.
- Tumor de Wilms diagnosticado antes de los 2 años.
Para obtener una explicación completa de los efectos tardíos del tratamiento del cáncer en niños y adolescentes, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Referencias:
- Howlader N, Noone AM, Krapcho M, et al.: SEER Cancer Statistics Review (CSR) 1975-2016. Bethesda, Md: National Cancer Institute, 2019. Available online. Last accessed August 8, 2022.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
- Breslow N, Olshan A, Beckwith JB, et al.: Ethnic variation in the incidence, diagnosis, prognosis, and follow-up of children with Wilms' tumor. J Natl Cancer Inst 86 (1): 49-51, 1994.
- Breslow N, Olshan A, Beckwith JB, et al.: Epidemiology of Wilms tumor. Med Pediatr Oncol 21 (3): 172-81, 1993.
- Horner MJ, Ries LA, Krapcho M, et al.: SEER Cancer Statistics Review, 1975-2006. National Cancer Institute, 2009. Also available online. Last accessed August 21, 2023.
- Scott RH, Stiller CA, Walker L, et al.: Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J Med Genet 43 (9): 705-15, 2006.
- Narod SA, Hawkins MM, Robertson CM, et al.: Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet 60 (3): 474-85, 1997.
- Vujanić GM, Apps JR, Moroz V, et al.: Nephrogenic rests in Wilms tumors treated with preoperative chemotherapy: The UK SIOP Wilms Tumor 2001 Trial experience. Pediatr Blood Cancer 64 (11): , 2017.
- Dumoucel S, Gauthier-Villars M, Stoppa-Lyonnet D, et al.: Malformations, genetic abnormalities, and Wilms tumor. Pediatr Blood Cancer 61 (1): 140-4, 2014.
- Gracia Bouthelier R, Lapunzina P: Follow-up and risk of tumors in overgrowth syndromes. J Pediatr Endocrinol Metab 18 (Suppl 1): 1227-35, 2005.
- Lapunzina P: Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet 137 (1): 53-71, 2005.
- Treger TD, Chowdhury T, Pritchard-Jones K, et al.: The genetic changes of Wilms tumour. Nat Rev Nephrol 15 (4): 240-251, 2019.
- Duffy KA, Trout KL, Gunckle JM, et al.: Results From the WAGR Syndrome Patient Registry: Characterization of WAGR Spectrum and Recommendations for Care Management. Front Pediatr 9: 733018, 2021.
- Clericuzio CL: Clinical phenotypes and Wilms tumor. Med Pediatr Oncol 21 (3): 182-7, 1993.
- Fischbach BV, Trout KL, Lewis J, et al.: WAGR syndrome: a clinical review of 54 cases. Pediatrics 116 (4): 984-8, 2005.
- Breslow NE, Norris R, Norkool PA, et al.: Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: a report from the National Wilms Tumor Study Group. J Clin Oncol 21 (24): 4579-85, 2003.
- Hol JA, Jongmans MCJ, Sudour-Bonnange H, et al.: Clinical characteristics and outcomes of children with WAGR syndrome and Wilms tumor and/or nephroblastomatosis: The 30-year SIOP-RTSG experience. Cancer 127 (4): 628-638, 2021.
- Barbosa AS, Hadjiathanasiou CG, Theodoridis C, et al.: The same mutation affecting the splicing of WT1 gene is present on Frasier syndrome patients with or without Wilms' tumor. Hum Mutat 13 (2): 146-53, 1999.
- Koziell AB, Grundy R, Barratt TM, et al.: Evidence for the genetic heterogeneity of nephropathic phenotypes associated with Denys-Drash and Frasier syndromes. Am J Hum Genet 64 (6): 1778-81, 1999.
- Royer-Pokora B, Beier M, Henzler M, et al.: Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 127 (3): 249-57, 2004.
- Pelletier J, Bruening W, Kashtan CE, et al.: Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67 (2): 437-47, 1991.
- Mueller RF: The Denys-Drash syndrome. J Med Genet 31 (6): 471-7, 1994.
- Barbaux S, Niaudet P, Gubler MC, et al.: Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet 17 (4): 467-70, 1997.
- Connolly GK, Harris RD, Shumate C, et al.: Pediatric cancer incidence among individuals with overgrowth syndromes and overgrowth features: A population-based assessment in seven million children. Cancer 130 (3): 467-475, 2024.
- Porteus MH, Narkool P, Neuberg D, et al.: Characteristics and outcome of children with Beckwith-Wiedemann syndrome and Wilms' tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 18 (10): 2026-31, 2000.
- Rump P, Zeegers MP, van Essen AJ: Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 136 (1): 95-104, 2005.
- Choufani S, Shuman C, Weksberg R: Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet 163C (2): 131-40, 2013.
- Eggermann T, Algar E, Lapunzina P, et al.: Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur J Hum Genet 22 (3): , 2014.
- Ibrahim A, Kirby G, Hardy C, et al.: Methylation analysis and diagnostics of Beckwith-Wiedemann syndrome in 1,000 subjects. Clin Epigenetics 6 (1): 11, 2014.
- Brioude F, Lacoste A, Netchine I, et al.: Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 80 (6): 457-65, 2013.
- Mussa A, Russo S, Larizza L, et al.: (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: a paradigm for genomic medicine. Clin Genet 89 (4): 403-415, 2016.
- Green DM, Breslow NE, Beckwith JB, et al.: Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol 21 (3): 188-92, 1993.
- DeBaun MR, Siegel MJ, Choyke PL: Nephromegaly in infancy and early childhood: a risk factor for Wilms tumor in Beckwith-Wiedemann syndrome. J Pediatr 132 (3 Pt 1): 401-4, 1998.
- DeBaun MR, Tucker MA: Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 132 (3 Pt 1): 398-400, 1998.
- Hol JA, Kuiper RP, van Dijk F, et al.: Prevalence of (Epi)genetic Predisposing Factors in a 5-Year Unselected National Wilms Tumor Cohort: A Comprehensive Clinical and Genomic Characterization. J Clin Oncol 40 (17): 1892-1902, 2022.
- Milani D, Pezzani L, Tabano S, et al.: Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet 7: 169-75, 2014.
- Morris MR, Astuti D, Maher ER: Perlman syndrome: overgrowth, Wilms tumor predisposition and DIS3L2. Am J Med Genet C Semin Med Genet 163C (2): 106-13, 2013.
- Astuti D, Morris MR, Cooper WN, et al.: Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44 (3): 277-84, 2012.
- Golabi M, Leung A, Lopez C: Simpson-Golabi-Behmel Syndrome Type 1. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed December 22, 2022.
- Peterman CM, Fevurly RD, Alomari AI, et al.: Sonographic screening for Wilms tumor in children with CLOVES syndrome. Pediatr Blood Cancer 64 (12): , 2017.
- Fagali C, Kok F, Nicola P, et al.: MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet 52 (5): 333-6, 2009 Sep-Oct.
- Isidor B, Bourdeaut F, Lafon D, et al.: Wilms' tumor in patients with 9q22.3 microdeletion syndrome suggests a role for PTCH1 in nephroblastomas. Eur J Hum Genet 21 (7): 784-7, 2013.
- Cayrol J, Nightingale M, Challis J, et al.: Wilms Tumor Associated With the 9q22.3 Microdeletion Syndrome: 2 New Case Reports and a Review of The Literature. J Pediatr Hematol Oncol 41 (8): e517-e520, 2019.
- Cairney AE, Andrews M, Greenberg M, et al.: Wilms tumor in three patients with Bloom syndrome. J Pediatr 111 (3): 414-6, 1987.
- Hartley AL, Birch JM, Tricker K, et al.: Wilms' tumor in the Li-Fraumeni cancer family syndrome. Cancer Genet Cytogenet 67 (2): 133-5, 1993.
- Bourdeaut F, Guiochon-Mantel A, Fabre M, et al.: Alagille syndrome and nephroblastoma: Unusual coincidence of two rare disorders. Pediatr Blood Cancer 50 (4): 908-11, 2008.
- Russell B, Johnston JJ, Biesecker LG, et al.: Clinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet A 167A (9): 2122-31, 2015.
- Bonaïti-Pellié C, Chompret A, Tournade MF, et al.: Genetics and epidemiology of Wilms' tumor: the French Wilms' tumor study. Med Pediatr Oncol 20 (4): 284-91, 1992.
- Winther JF, Sankila R, Boice JD, et al.: Cancer in siblings of children with cancer in the Nordic countries: a population-based cohort study. Lancet 358 (9283): 711-7, 2001.
- Breslow NE, Olson J, Moksness J, et al.: Familial Wilms' tumor: a descriptive study. Med Pediatr Oncol 27 (5): 398-403, 1996.
- Li FP, Williams WR, Gimbrere K, et al.: Heritable fraction of unilateral Wilms tumor. Pediatrics 81 (1): 147-9, 1988.
- Rahman N, Arbour L, Tonin P, et al.: Evidence for a familial Wilms' tumour gene (FWT1) on chromosome 17q12-q21. Nat Genet 13 (4): 461-3, 1996.
- McDonald JM, Douglass EC, Fisher R, et al.: Linkage of familial Wilms' tumor predisposition to chromosome 19 and a two-locus model for the etiology of familial tumors. Cancer Res 58 (7): 1387-90, 1998.
- Halliday BJ, Fukuzawa R, Markie DM, et al.: Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PLoS Genet 14 (6): e1007399, 2018.
- Grundy P, Koufos A, Morgan K, et al.: Familial predisposition to Wilms' tumour does not map to the short arm of chromosome 11. Nature 336 (6197): 374-6, 1988.
- Little SE, Hanks SP, King-Underwood L, et al.: Frequency and heritability of WT1 mutations in nonsyndromic Wilms' tumor patients: a UK Children's Cancer Study Group Study. J Clin Oncol 22 (20): 4140-6, 2004.
- Hanks S, Perdeaux ER, Seal S, et al.: Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun 5: 4398, 2014.
- Scott RH, Douglas J, Baskcomb L, et al.: Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet 40 (11): 1329-34, 2008.
- Grønskov K, Olsen JH, Sand A, et al.: Population-based risk estimates of Wilms tumor in sporadic aniridia. A comprehensive mutation screening procedure of PAX6 identifies 80% of mutations in aniridia. Hum Genet 109 (1): 11-8, 2001.
- Clericuzio C, Hingorani M, Crolla JA, et al.: Clinical utility gene card for: WAGR syndrome. Eur J Hum Genet 19 (4): , 2011.
- Kalish JM, Biesecker LG, Brioude F, et al.: Nomenclature and definition in asymmetric regional body overgrowth. Am J Med Genet A 173 (7): 1735-1738, 2017.
- Shuman C, Smith AC, Steele L, et al.: Constitutional UPD for chromosome 11p15 in individuals with isolated hemihyperplasia is associated with high tumor risk and occurs following assisted reproductive technologies. Am J Med Genet A 140 (14): 1497-503, 2006.
- Shanske AL: Trisomy 18 in a second 20-year-old woman. Am J Med Genet A 140 (9): 966-7, 2006.
- Reid S, Renwick A, Seal S, et al.: Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 42 (2): 147-51, 2005.
- Hirsch B, Shimamura A, Moreau L, et al.: Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 103 (7): 2554-9, 2004.
- Reid S, Schindler D, Hanenberg H, et al.: Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39 (2): 162-4, 2007.
- Rios P, Bauer H, Schleiermacher G, et al.: Environmental exposures related to parental habits in the perinatal period and the risk of Wilms' tumor in children. Cancer Epidemiol 66: 101706, 2020.
- Coorens THH, Treger TD, Al-Saadi R, et al.: Embryonal precursors of Wilms tumor. Science 366 (6470): 1247-1251, 2019.
- Gadd S, Huff V, Walz AL, et al.: A Children's Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat Genet 49 (10): 1487-1494, 2017.
- Wegert J, Wittmann S, Leuschner I, et al.: WTX inactivation is a frequent, but late event in Wilms tumors without apparent clinical impact. Genes Chromosomes Cancer 48 (12): 1102-11, 2009.
- Ruteshouser EC, Robinson SM, Huff V: Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47 (6): 461-70, 2008.
- Walz AL, Ooms A, Gadd S, et al.: Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell 27 (2): 286-97, 2015.
- Wegert J, Ishaque N, Vardapour R, et al.: Mutations in the SIX1/2 pathway and the DROSHA/DGCR8 miRNA microprocessor complex underlie high-risk blastemal type Wilms tumors. Cancer Cell 27 (2): 298-311, 2015.
- Rakheja D, Chen KS, Liu Y, et al.: Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2: 4802, 2014.
- Torrezan GT, Ferreira EN, Nakahata AM, et al.: Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun 5: 4039, 2014.
- Dome JS, Huff V: Wilms Tumor Predisposition. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed August 16, 2022.
- Mahamdallie SS, Hanks S, Karlin KL, et al.: Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet 47 (12): 1471-4, 2015.
- Huff V: Wilms tumor genetics. Am J Med Genet 79 (4): 260-7, 1998.
- Scott RH, Murray A, Baskcomb L, et al.: Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 3 (3): 327-35, 2012.
- Corbin M, de Reyniès A, Rickman DS, et al.: WNT/beta-catenin pathway activation in Wilms tumors: a unifying mechanism with multiple entries? Genes Chromosomes Cancer 48 (9): 816-27, 2009.
- Maiti S, Alam R, Amos CI, et al.: Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res 60 (22): 6288-92, 2000.
- Gadd S, Huff V, Huang CC, et al.: Clinically relevant subsets identified by gene expression patterns support a revised ontogenic model of Wilms tumor: a Children's Oncology Group Study. Neoplasia 14 (8): 742-56, 2012.
- Breslow NE, Beckwith JB, Perlman EJ, et al.: Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 47 (3): 260-7, 2006.
- Fukuzawa R, Heathcott RW, More HE, et al.: Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: two case studies. J Clin Pathol 60 (9): 1013-6, 2007.
- Perlman EJ, Gadd S, Arold ST, et al.: MLLT1 YEATS domain mutations in clinically distinctive Favourable Histology Wilms tumours. Nat Commun 6: 10013, 2015.
- Diller L, Ghahremani M, Morgan J, et al.: Constitutional WT1 mutations in Wilms' tumor patients. J Clin Oncol 16 (11): 3634-40, 1998.
- Perlman EJ, Grundy PE, Anderson JR, et al.: WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk wilms tumors treated with surgery alone: a children's oncology group study. J Clin Oncol 29 (6): 698-703, 2011.
- Lipska BS, Ranchin B, Iatropoulos P, et al.: Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int 85 (5): 1169-78, 2014.
- Lehnhardt A, Karnatz C, Ahlenstiel-Grunow T, et al.: Clinical and molecular characterization of patients with heterozygous mutations in wilms tumor suppressor gene 1. Clin J Am Soc Nephrol 10 (5): 825-31, 2015.
- Marakhonov AV, Vasilyeva TA, Voskresenskaya AA, et al.: LMO2 gene deletions significantly worsen the prognosis of Wilms' tumor development in patients with WAGR syndrome. Hum Mol Genet 28 (19): 3323-3326, 2019.
- Scott RH, Walker L, Olsen ØE, et al.: Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 91 (12): 995-9, 2006.
- Koesters R, Ridder R, Kopp-Schneider A, et al.: Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms' tumors. Cancer Res 59 (16): 3880-2, 1999.
- Koesters R, Niggli F, von Knebel Doeberitz M, et al.: Nuclear accumulation of beta-catenin protein in Wilms' tumours. J Pathol 199 (1): 68-76, 2003.
- Major MB, Camp ND, Berndt JD, et al.: Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science 316 (5827): 1043-6, 2007.
- Rivera MN, Kim WJ, Wells J, et al.: An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 315 (5812): 642-5, 2007.
- Fukuzawa R, Anaka MR, Weeks RJ, et al.: Canonical WNT signalling determines lineage specificity in Wilms tumour. Oncogene 28 (8): 1063-75, 2009.
- Jenkins ZA, van Kogelenberg M, Morgan T, et al.: Germline mutations in WTX cause a sclerosing skeletal dysplasia but do not predispose to tumorigenesis. Nat Genet 41 (1): 95-100, 2009.
- Grohmann A, Tanneberger K, Alzner A, et al.: AMER1 regulates the distribution of the tumor suppressor APC between microtubules and the plasma membrane. J Cell Sci 120 (Pt 21): 3738-47, 2007.
- Satoh Y, Nakadate H, Nakagawachi T, et al.: Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer 95 (4): 541-7, 2006.
- Algar EM, St Heaps L, Darmanian A, et al.: Paternally inherited submicroscopic duplication at 11p15.5 implicates insulin-like growth factor II in overgrowth and Wilms' tumorigenesis. Cancer Res 67 (5): 2360-5, 2007.
- Lennerz JK, Timmerman RJ, Grange DK, et al.: Addition of H19 'loss of methylation testing' for Beckwith-Wiedemann syndrome (BWS) increases the diagnostic yield. J Mol Diagn 12 (5): 576-88, 2010.
- Mussa A, Molinatto C, Baldassarre G, et al.: Cancer Risk in Beckwith-Wiedemann Syndrome: A Systematic Review and Meta-Analysis Outlining a Novel (Epi)Genotype Specific Histotype Targeted Screening Protocol. J Pediatr 176: 142-149.e1, 2016.
- Bliek J, Gicquel C, Maas S, et al.: Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J Pediatr 145 (6): 796-9, 2004.
- Bjornsson HT, Brown LJ, Fallin MD, et al.: Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst 99 (16): 1270-3, 2007.
- Fukuzawa R, Breslow NE, Morison IM, et al.: Epigenetic differences between Wilms' tumours in white and east-Asian children. Lancet 363 (9407): 446-51, 2004.
- Gratias EJ, Dome JS, Jennings LJ, et al.: Association of Chromosome 1q Gain With Inferior Survival in Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group. J Clin Oncol 34 (26): 3189-94, 2016.
- Chagtai T, Zill C, Dainese L, et al.: Gain of 1q As a Prognostic Biomarker in Wilms Tumors (WTs) Treated With Preoperative Chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 Trial: A SIOP Renal Tumours Biology Consortium Study. J Clin Oncol 34 (26): 3195-203, 2016.
- Gadd S, Huff V, Skol AD, et al.: Genetic changes associated with relapse in favorable histology Wilms tumor: A Children's Oncology Group AREN03B2 study. Cell Rep Med 3 (6): 100644, 2022.
- Grundy PE, Breslow NE, Li S, et al.: Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol 23 (29): 7312-21, 2005.
- Messahel B, Williams R, Ridolfi A, et al.: Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study. Eur J Cancer 45 (5): 819-26, 2009.
- Spreafico F, Gamba B, Mariani L, et al.: Loss of heterozygosity analysis at different chromosome regions in Wilms tumor confirms 1p allelic loss as a marker of worse prognosis: a study from the Italian Association of Pediatric Hematology and Oncology. J Urol 189 (1): 260-6, 2013.
- Gratias EJ, Jennings LJ, Anderson JR, et al.: Gain of 1q is associated with inferior event-free and overall survival in patients with favorable histology Wilms tumor: a report from the Children's Oncology Group. Cancer 119 (21): 3887-94, 2013.
- Hohenstein P, Pritchard-Jones K, Charlton J: The yin and yang of kidney development and Wilms' tumors. Genes Dev 29 (5): 467-82, 2015.
- Foulkes WD, Priest JR, Duchaine TF: DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 14 (10): 662-72, 2014.
- Wu MK, Sabbaghian N, Xu B, et al.: Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 230 (2): 154-64, 2013.
- Palculict TB, Ruteshouser EC, Fan Y, et al.: Identification of germline DICER1 mutations and loss of heterozygosity in familial Wilms tumour. J Med Genet 53 (6): 385-8, 2016.
- Chang HM, Triboulet R, Thornton JE, et al.: A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature 497 (7448): 244-8, 2013.
- Alessandri JL, Cuillier F, Ramful D, et al.: Perlman syndrome: report, prenatal findings and review. Am J Med Genet A 146A (19): 2532-7, 2008.
- Bardeesy N, Falkoff D, Petruzzi MJ, et al.: Anaplastic Wilms' tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 7 (1): 91-7, 1994.
- el Bahtimi R, Hazen-Martin DJ, Re GG, et al.: Immunophenotype, mRNA expression, and gene structure of p53 in Wilms' tumors. Mod Pathol 9 (3): 238-44, 1996.
- Wallkamm V, Dörlich R, Rahm K, et al.: Live imaging of Xwnt5A-ROR2 complexes. PLoS One 9 (10): e109428, 2014.
- Ooms AH, Gadd S, Gerhard DS, et al.: Significance of TP53 Mutation in Wilms Tumors with Diffuse Anaplasia: A Report from the Children's Oncology Group. Clin Cancer Res 22 (22): 5582-5591, 2016.
- Williams RD, Al-Saadi R, Chagtai T, et al.: Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms' tumor. Clin Cancer Res 16 (7): 2036-45, 2010.
- Mahamdallie S, Yost S, Poyastro-Pearson E, et al.: Identification of new Wilms tumour predisposition genes: an exome sequencing study. Lancet Child Adolesc Health 3 (5): 322-331, 2019.
- Armstrong AE, Gadd S, Huff V, et al.: A unique subset of low-risk Wilms tumors is characterized by loss of function of TRIM28 (KAP1), a gene critical in early renal development: A Children's Oncology Group study. PLoS One 13 (12): e0208936, 2018.
- Diets IJ, Hoyer J, Ekici AB, et al.: TRIM28 haploinsufficiency predisposes to Wilms tumor. Int J Cancer 145 (4): 941-951, 2019.
- Hol JA, Diets IJ, de Krijger RR, et al.: TRIM28 variants and Wilms' tumour predisposition. J Pathol 254 (4): 494-504, 2021.
- Garavelli L, Piemontese MR, Cavazza A, et al.: Multiple tumor types including leiomyoma and Wilms tumor in a patient with Gorlin syndrome due to 9q22.3 microdeletion encompassing the PTCH1 and FANC-C loci. Am J Med Genet A 161A (11): 2894-901, 2013.
- Cajaiba MM, Bale AE, Alvarez-Franco M, et al.: Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nat Clin Pract Oncol 3 (10): 575-80, 2006.
- Williams RD, Chagtai T, Alcaide-German M, et al.: Multiple mechanisms of MYCN dysregulation in Wilms tumour. Oncotarget 6 (9): 7232-43, 2015.
- Fievet A, Belaud-Rotureau MA, Dugay F, et al.: Involvement of germline DDX1-MYCN duplication in inherited nephroblastoma. Eur J Med Genet 56 (12): 643-7, 2013.
- Jiménez Martín O, Schlosser A, Furtwängler R, et al.: MYCN and MAX alterations in Wilms tumor and identification of novel N-MYC interaction partners as biomarker candidates. Cancer Cell Int 21 (1): 555, 2021.
- Martins AG, Pinto AT, Domingues R, et al.: Identification of a novel CTR9 germline mutation in a family with Wilms tumor. Eur J Med Genet 61 (5): 294-299, 2018.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- Argani P, Tickoo SK, Matoso A, et al.: Adult Wilms Tumor: Genetic Evidence of Origin of a Subset of Cases From Metanephric Adenoma. Am J Surg Pathol 46 (7): 988-999, 2022.
- Choueiri TK, Cheville J, Palescandolo E, et al.: BRAF mutations in metanephric adenoma of the kidney. Eur Urol 62 (5): 917-22, 2012.
- Wobker SE, Matoso A, Pratilas CA, et al.: Metanephric Adenoma-Epithelial Wilms Tumor Overlap Lesions: An Analysis of BRAF Status. Am J Surg Pathol 43 (9): 1157-1169, 2019.
- Charlton J, Irtan S, Bergeron C, et al.: Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev Mol Med 19: e8, 2017.
- Beckwith JB, Kiviat NB, Bonadio JF: Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol 10 (1-2): 1-36, 1990.
- Hu M, Fletcher J, McCahon E, et al.: Bilateral Wilms tumor and early presentation in pediatric patients is associated with the truncation of the Wilms tumor 1 protein. J Pediatr 163 (1): 224-9, 2013.
- Murphy AJ, Davidoff AM: Bilateral Wilms Tumor: A Surgical Perspective. Children (Basel) 5 (10): , 2018.
- Kalish JM, Doros L, Helman LJ, et al.: Surveillance Recommendations for Children with Overgrowth Syndromes and Predisposition to Wilms Tumors and Hepatoblastoma. Clin Cancer Res 23 (13): e115-e122, 2017.
- Mussa A, Duffy KA, Carli D, et al.: The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol 145 (12): 3115-3123, 2019.
- Teplick A, Kowalski M, Biegel JA, et al.: Educational paper: screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr 170 (3): 285-94, 2011.
- Hol JA, Jewell R, Chowdhury T, et al.: Wilms tumour surveillance in at-risk children: Literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer 153: 51-63, 2021.
- Ehrlich PF, Chi YY, Chintagumpala MM, et al.: Results of Treatment for Patients With Multicentric or Bilaterally Predisposed Unilateral Wilms Tumor (AREN0534): A report from the Children's Oncology Group. Cancer 126 (15): 3516-3525, 2020.
- Brioude F, Kalish JM, Mussa A, et al.: Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol 14 (4): 229-249, 2018.
- Lima Cunha D, Arno G, Corton M, et al.: The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes (Basel) 10 (12): , 2019.
- Hingorani M, Hanson I, van Heyningen V: Aniridia. Eur J Hum Genet 20 (10): 1011-7, 2012.
- van Heyningen V, Hoovers JM, de Kraker J, et al.: Raised risk of Wilms tumour in patients with aniridia and submicroscopic WT1 deletion. J Med Genet 44 (12): 787-90, 2007.
- Russell B, Tan W-H, Graham JM: Bohring-Opitz Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed November 9, 2022.
- Fernandes C, Paúl A, Venâncio MM, et al.: Simpson-Golabi-Behmel syndrome: One family, same mutation, different outcome. Am J Med Genet A 185 (8): 2502-2506, 2021.
- Greene AK, Kieran M, Burrows PE, et al.: Wilms tumor screening is unnecessary in Klippel-Trenaunay syndrome. Pediatrics 113 (4): e326-9, 2004.
- Schultz KAP, Williams GM, Kamihara J, et al.: DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin Cancer Res 24 (10): 2251-2261, 2018.
- Mitchell SG, Pencheva B, Porter CC: Germline Genetics and Childhood Cancer: Emerging Cancer Predisposition Syndromes and Psychosocial Impacts. Curr Oncol Rep 21 (10): 85, 2019.
- Schoettler PJ, Smith CC, Nishitani M, et al.: Anaplastic sarcoma of the kidney (DICER1-sarcoma of the kidney): A report from the International Pleuropulmonary Blastoma/DICER1 Registry. Pediatr Blood Cancer 71 (8): e31090, 2024.
- Maciaszek JL, Oak N, Nichols KE: Recent advances in Wilms' tumor predisposition. Hum Mol Genet 29 (R2): R138-R149, 2020.
- Cullinan N, Villani A, Mourad S, et al.: An eHealth decision-support tool to prioritize referral practices for genetic evaluation of patients with Wilms tumor. Int J Cancer 146 (4): 1010-1017, 2020.
- Green DM: Wilms' tumor. In: Greem DM: Diagnosis and Management of Malignant Solid Tumors in Infants and Children. Martinus Nijhoff Publishing, 1985, pp 129-86.
- Artunduaga M, Eklund M, van der Beek JN, et al.: Imaging of pediatric renal tumors: A COG Diagnostic Imaging Committee/SPR Oncology Committee White Paper focused on Wilms tumor and nephrogenic rests. Pediatr Blood Cancer 70 (Suppl 4): e30004, 2023.
- Khanna G, Naranjo A, Hoffer F, et al.: Detection of preoperative wilms tumor rupture with CT: a report from the Children's Oncology Group. Radiology 266 (2): 610-7, 2013.
- McDonald K, Duffy P, Chowdhury T, et al.: Added value of abdominal cross-sectional imaging (CT or MRI) in staging of Wilms' tumours. Clin Radiol 68 (1): 16-20, 2013.
- Ritchey ML, Shamberger RC, Hamilton T, et al.: Fate of bilateral renal lesions missed on preoperative imaging: a report from the National Wilms Tumor Study Group. J Urol 174 (4 Pt 2): 1519-21; discussion 1521, 2005.
- Khanna G, Rosen N, Anderson JR, et al.: Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: a report from the Children's Oncology Group. Pediatr Blood Cancer 58 (4): 551-5, 2012.
- Sandberg JK, Chi YY, Smith EA, et al.: Imaging Characteristics of Nephrogenic Rests Versus Small Wilms Tumors: A Report From the Children's Oncology Group Study AREN03B2. AJR Am J Roentgenol 214 (5): 987-994, 2020.
- Watson T, Oostveen M, Rogers H, et al.: The role of imaging in the initial investigation of paediatric renal tumours. Lancet Child Adolesc Health 4 (3): 232-241, 2020.
- Servaes SE, Hoffer FA, Smith EA, et al.: Imaging of Wilms tumor: an update. Pediatr Radiol 49 (11): 1441-1452, 2019.
- Al-Hadidi A, Rinehardt HN, Sutthatarn P, et al.: Incidence and management of pleural effusions in patients with Wilms tumor: A Pediatric Surgical Oncology Research Collaborative study. Int J Cancer 151 (10): 1696-1702, 2022.
- Begent J, Sebire NJ, Levitt G, et al.: Pilot study of F(18)-Fluorodeoxyglucose Positron Emission Tomography/computerised tomography in Wilms' tumour: correlation with conventional imaging, pathology and immunohistochemistry. Eur J Cancer 47 (3): 389-96, 2011.
- Callaghan MU, Wong TE, Federici AB: Treatment of acquired von Willebrand syndrome in childhood. Blood 122 (12): 2019-22, 2013.
- Charlebois J, Rivard GÉ, St-Louis J: Management of acquired von Willebrand syndrome. Transfus Apher Sci 57 (6): 721-723, 2018.
- Shamberger RC, Guthrie KA, Ritchey ML, et al.: Surgery-related factors and local recurrence of Wilms tumor in National Wilms Tumor Study 4. Ann Surg 229 (2): 292-7, 1999.
- Hamilton TE, Green DM, Perlman EJ, et al.: Bilateral Wilms' tumor with anaplasia: lessons from the National Wilms' Tumor Study. J Pediatr Surg 41 (10): 1641-4, 2006.
- Servaes S, Khanna G, Naranjo A, et al.: Comparison of diagnostic performance of CT and MRI for abdominal staging of pediatric renal tumors: a report from the Children's Oncology Group. Pediatr Radiol 45 (2): 166-72, 2015.
- Ehrlich P, Chi YY, Chintagumpala MM, et al.: Results of the First Prospective Multi-institutional Treatment Study in Children With Bilateral Wilms Tumor (AREN0534): A Report From the Children's Oncology Group. Ann Surg 266 (3): 470-478, 2017.
- Othersen HB, DeLorimer A, Hrabovsky E, et al.: Surgical evaluation of lymph node metastases in Wilms' tumor. J Pediatr Surg 25 (3): 330-1, 1990.
- Shamberger RC, Ritchey ML, Haase GM, et al.: Intravascular extension of Wilms tumor. Ann Surg 234 (1): 116-21, 2001.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Green DM, Breslow NE, Beckwith JB, et al.: Effect of duration of treatment on treatment outcome and cost of treatment for Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (12): 3744-51, 1998.
- Kalapurakal JA, Dome JS, Perlman EJ, et al.: Management of Wilms' tumour: current practice and future goals. Lancet Oncol 5 (1): 37-46, 2004.
- Ehrlich PF: Wilms tumor: progress and considerations for the surgeon. Surg Oncol 16 (3): 157-71, 2007.
- Dome JS, Cotton CA, Perlman EJ, et al.: Treatment of anaplastic histology Wilms' tumor: results from the fifth National Wilms' Tumor Study. J Clin Oncol 24 (15): 2352-8, 2006.
- Shamberger RC, Anderson JR, Breslow NE, et al.: Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg 251 (3): 555-8, 2010.
- Fernandez CV, Perlman EJ, Mullen EA, et al.: Clinical Outcome and Biological Predictors of Relapse After Nephrectomy Only for Very Low-risk Wilms Tumor: A Report From Children's Oncology Group AREN0532. Ann Surg 265 (4): 835-840, 2017.
- Hol JA, Lopez-Yurda MI, Van Tinteren H, et al.: Prognostic significance of age in 5631 patients with Wilms tumour prospectively registered in International Society of Paediatric Oncology (SIOP) 93-01 and 2001. PLoS One 14 (8): e0221373, 2019.
- Qian DC, Sykes-Martin KD, Tobillo R, et al.: Impact of Age on Overall Survival Among Children With Wilms Tumor: A Population-based Registry Analysis. Am J Clin Oncol 46 (5): 213-218, 2023.
- Mitry E, Ciccolallo L, Coleman MP, et al.: Incidence of and survival from Wilms' tumour in adults in Europe: data from the EUROCARE study. Eur J Cancer 42 (14): 2363-8, 2006.
- Ali AN, Diaz R, Shu HK, et al.: A Surveillance, Epidemiology and End Results (SEER) program comparison of adult and pediatric Wilms' tumor. Cancer 118 (9): 2541-51, 2012.
- Walker JP, Saltzman AF, Kessler ER, et al.: Adult Wilms Tumor During Pregnancy: Case Report and Literature Review. Urology 129: 200-205, 2019.
- Brown JT, Harik LR, Barbee MS, et al.: Multidisciplinary Care of Adult Wilms' Tumor During Pregnancy: A Case Report and Review of the Literature. Clin Genitourin Cancer 18 (1): e1-e4, 2020.
- Chen I, Pasalic D, Fischer-Valuck B, et al.: Disparity in Outcomes for Adolescent and Young Adult Patients Diagnosed With Pediatric Solid Tumors Across 4 Decades. Am J Clin Oncol 41 (5): 471-475, 2018.
- Saltzman AF, Carrasco A, Amini A, et al.: Patterns of Care and Survival Comparison of Adult and Pediatric Wilms Tumor in the United States: A Study of the National Cancer Database. Urology 135: 50-56, 2020.
- Kalapurakal JA, Nan B, Norkool P, et al.: Treatment outcomes in adults with favorable histologic type Wilms tumor-an update from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 60 (5): 1379-84, 2004.
- Arrigo S, Beckwith JB, Sharples K, et al.: Better survival after combined modality care for adults with Wilms' tumor. A report from the National Wilms' Tumor Study. Cancer 66 (5): 827-30, 1990.
- Byrd RL, Evans AE, D'Angio GJ: Adult Wilms tumor: effect of combined therapy on survival. J Urol 127 (4): 648-51, 1982.
- de Vries-Brilland M, Sionneau B, Dutriaux C, et al.: Successful Treatment of Metastatic Adult Wilms Tumor With Anti-BRAF Treatment: A Case Report and a Brief Review of the Literature. Clin Genitourin Cancer 17 (4): e721-e723, 2019.
- Segers H, van den Heuvel-Eibrink MM, Pritchard-Jones K, et al.: Management of adults with Wilms' tumor: recommendations based on international consensus. Expert Rev Anticancer Ther 11 (7): 1105-13, 2011.
- Perlman EJ: Pediatric renal tumors: practical updates for the pathologist. Pediatr Dev Pathol 8 (3): 320-38, 2005 May-Jun.
- Parsons LN, Mullen EA, Geller JI, et al.: Outcome analysis of stage I epithelial-predominant favorable-histology Wilms tumors: A report from Children's Oncology Group study AREN03B2. Cancer 126 (12): 2866-2871, 2020.
- Popov SD, Sebire NJ, Pritchard-Jones K, et al.: Renal tumors in children aged 10-16 Years: a report from the United Kingdom Children's Cancer and Leukaemia Group. Pediatr Dev Pathol 14 (3): 189-93, 2011 May-Jun.
- Indolfi P, Jenkner A, Terenziani M, et al.: Synchronous bilateral Wilms tumor: a report from the Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP). Cancer 119 (8): 1586-92, 2013.
- Hamilton TE, Ritchey ML, Haase GM, et al.: The management of synchronous bilateral Wilms tumor: a report from the National Wilms Tumor Study Group. Ann Surg 253 (5): 1004-10, 2011.
- Williams RD, Al-Saadi R, Natrajan R, et al.: Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer 50 (12): 982-95, 2011.
- Vujanić GM, Harms D, Sandstedt B, et al.: New definitions of focal and diffuse anaplasia in Wilms tumor: the International Society of Paediatric Oncology (SIOP) experience. Med Pediatr Oncol 32 (5): 317-23, 1999.
- Faria P, Beckwith JB, Mishra K, et al.: Focal versus diffuse anaplasia in Wilms tumor--new definitions with prognostic significance: a report from the National Wilms Tumor Study Group. Am J Surg Pathol 20 (8): 909-20, 1996.
- Beckwith JB: Precursor lesions of Wilms tumor: clinical and biological implications. Med Pediatr Oncol 21 (3): 158-68, 1993.
- Hennigar RA, O'Shea PA, Grattan-Smith JD: Clinicopathologic features of nephrogenic rests and nephroblastomatosis. Adv Anat Pathol 8 (5): 276-89, 2001.
- Fukuzawa R, Reeve AE: Molecular pathology and epidemiology of nephrogenic rests and Wilms tumors. J Pediatr Hematol Oncol 29 (9): 589-94, 2007.
- Vuononvirta R, Sebire NJ, Dallosso AR, et al.: Perilobar nephrogenic rests are nonobligate molecular genetic precursor lesions of insulin-like growth factor-II-associated Wilms tumors. Clin Cancer Res 14 (23): 7635-44, 2008.
- Perlman EJ, Faria P, Soares A, et al.: Hyperplastic perilobar nephroblastomatosis: long-term survival of 52 patients. Pediatr Blood Cancer 46 (2): 203-21, 2006.
- Lange J, Peterson SM, Takashima JR, et al.: Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor. J Urol 186 (2): 378-86, 2011.
- Coppes MJ, Arnold M, Beckwith JB, et al.: Factors affecting the risk of contralateral Wilms tumor development: a report from the National Wilms Tumor Study Group. Cancer 85 (7): 1616-25, 1999.
- Cooke A, Deshpande AV, La Hei ER, et al.: Ectopic nephrogenic rests in children: the clinicosurgical implications. J Pediatr Surg 44 (12): e13-6, 2009.
- Wilms' tumor: status report, 1990. By the National Wilms' Tumor Study Committee. J Clin Oncol 9 (5): 877-87, 1991.
- Green DM, Breslow NE, D'Angio GJ, et al.: Outcome of patients with Stage II/favorable histology Wilms tumor with and without local tumor spill: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 61 (1): 134-9, 2014.
- Ehrlich PF, Anderson JR, Ritchey ML, et al.: Clinicopathologic findings predictive of relapse in children with stage III favorable-histology Wilms tumor. J Clin Oncol 31 (9): 1196-201, 2013.
- D'Angio GJ, Breslow N, Beckwith JB, et al.: Treatment of Wilms' tumor. Results of the Third National Wilms' Tumor Study. Cancer 64 (2): 349-60, 1989.
- Jereb B, Burgers JM, Tournade MF, et al.: Radiotherapy in the SIOP (International Society of Pediatric Oncology) nephroblastoma studies: a review. Med Pediatr Oncol 22 (4): 221-7, 1994.
- Green DM: The treatment of stages I-IV favorable histology Wilms' tumor. J Clin Oncol 22 (8): 1366-72, 2004.
- Graf N, Tournade MF, de Kraker J: The role of preoperative chemotherapy in the management of Wilms' tumor. The SIOP studies. International Society of Pediatric Oncology. Urol Clin North Am 27 (3): 443-54, 2000.
- Vujanić GM, D'Hooghe E, Popov SD, et al.: The effect of preoperative chemotherapy on histological subtyping and staging of Wilms tumors: The United Kingdom Children's Cancer Study Group (UKCCSG) Wilms tumor trial 3 (UKW3) experience. Pediatr Blood Cancer 66 (3): e27549, 2019.
- van den Heuvel-Eibrink MM, Hol JA, Pritchard-Jones K, et al.: Position paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat Rev Urol 14 (12): 743-752, 2017.
- Green DM, Breslow NE, Beckwith JB, et al.: Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 16 (1): 237-45, 1998.
- D'Angio GJ, Evans AE, Breslow N, et al.: The treatment of Wilms' tumor: Results of the national Wilms' tumor study. Cancer 38 (2): 633-46, 1976.
- D'Angio GJ, Evans A, Breslow N, et al.: The treatment of Wilms' tumor: results of the Second National Wilms' Tumor Study. Cancer 47 (9): 2302-11, 1981.
- Kieran K, Anderson JR, Dome JS, et al.: Lymph node involvement in Wilms tumor: results from National Wilms Tumor Studies 4 and 5. J Pediatr Surg 47 (4): 700-6, 2012.
- Ritchey M, Daley S, Shamberger RC, et al.: Ureteral extension in Wilms' tumor: a report from the National Wilms' Tumor Study Group (NWTSG). J Pediatr Surg 43 (9): 1625-9, 2008.
- Gow KW, Barnhart DC, Hamilton TE, et al.: Primary nephrectomy and intraoperative tumor spill: report from the Children's Oncology Group (COG) renal tumors committee. J Pediatr Surg 48 (1): 34-8, 2013.
- McNeil DE, Langer JC, Choyke P, et al.: Feasibility of partial nephrectomy for Wilms' tumor in children with Beckwith-Wiedemann syndrome who have been screened with abdominal ultrasonography. J Pediatr Surg 37 (1): 57-60, 2002.
- Scalabre A, Bergeron C, Brioude F, et al.: Is Nephron Sparing Surgery Justified in Wilms Tumor With Beckwith-Wiedemann Syndrome or Isolated Hemihypertrophy? Pediatr Blood Cancer 63 (9): 1571-7, 2016.
- Auber F, Jeanpierre C, Denamur E, et al.: Management of Wilms tumors in Drash and Frasier syndromes. Pediatr Blood Cancer 52 (1): 55-9, 2009.
- Neville H, Ritchey ML, Shamberger RC, et al.: The occurrence of Wilms tumor in horseshoe kidneys: a report from the National Wilms Tumor Study Group (NWTSG). J Pediatr Surg 37 (8): 1134-7, 2002.
- Ferrer FA, Rosen N, Herbst K, et al.: Image based feasibility of renal sparing surgery for very low risk unilateral Wilms tumors: a report from the Children's Oncology Group. J Urol 190 (5): 1846-51, 2013.
- Ritchey ML: Renal sparing surgery for Wilms tumor. J Urol 174 (4 Pt 1): 1172-3, 2005.
- Cozzi DA, Zani A: Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg 15 (1): 3-9, 2006.
- Ritchey ML, Kelalis PP, Breslow N, et al.: Surgical complications after nephrectomy for Wilms' tumor. Surg Gynecol Obstet 175 (6): 507-14, 1992.
- Ehrlich PF, Ferrer FA, Ritchey ML, et al.: Hepatic metastasis at diagnosis in patients with Wilms tumor is not an independent adverse prognostic factor for stage IV Wilms tumor: a report from the Children's Oncology Group/National Wilms Tumor Study Group. Ann Surg 250 (4): 642-8, 2009.
- Zhuge Y, Cheung MC, Yang R, et al.: Improved survival with lymph node sampling in Wilms tumor. J Surg Res 167 (2): e199-203, 2011.
- Ehrlich PF, Ritchey ML, Hamilton TE, et al.: Quality assessment for Wilms' tumor: a report from the National Wilms' Tumor Study-5. J Pediatr Surg 40 (1): 208-12; discussion 212-3, 2005.
- Fernandez CV, Mullen EA, Chi YY, et al.: Outcome and Prognostic Factors in Stage III Favorable-Histology Wilms Tumor: A Report From the Children's Oncology Group Study AREN0532. J Clin Oncol 36 (3): 254-261, 2018.
- Ritchey ML: Primary nephrectomy for Wilms' tumor: approach of the National Wilms' Tumor Study Group. Urology 47 (6): 787-91, 1996.
- Ritchey ML, Pringle KC, Breslow NE, et al.: Management and outcome of inoperable Wilms tumor. A report of National Wilms Tumor Study-3. Ann Surg 220 (5): 683-90, 1994.
- Ritchey ML, Shamberger RC, Haase G, et al.: Surgical complications after primary nephrectomy for Wilms' tumor: report from the National Wilms' Tumor Study Group. J Am Coll Surg 192 (1): 63-8; quiz 146, 2001.
- Naik-Mathuria B, Utria AF, Ehrlich PF, et al.: Management and Outcomes of Wilms Tumor With Suprarenal Intravascular Extension: A Pediatric Surgical Oncology Research Collaborative Study. Ann Surg 279 (3): 528-535, 2024.
- Tournade MF, Com-Nougué C, Voûte PA, et al.: Results of the Sixth International Society of Pediatric Oncology Wilms' Tumor Trial and Study: a risk-adapted therapeutic approach in Wilms' tumor. J Clin Oncol 11 (6): 1014-23, 1993.
- Oberholzer HF, Falkson G, De Jager LC: Successful management of inferior vena cava and right atrial nephroblastoma tumor thrombus with preoperative chemotherapy. Med Pediatr Oncol 20 (1): 61-3, 1992.
- Saarinen UM, Wikström S, Koskimies O, et al.: Percutaneous needle biopsy preceding preoperative chemotherapy in the management of massive renal tumors in children. J Clin Oncol 9 (3): 406-15, 1991.
- Dykes EH, Marwaha RK, Dicks-Mireaux C, et al.: Risks and benefits of percutaneous biopsy and primary chemotherapy in advanced Wilms' tumour. J Pediatr Surg 26 (5): 610-2, 1991.
- Thompson WR, Newman K, Seibel N, et al.: A strategy for resection of Wilms' tumor with vena cava or atrial extension. J Pediatr Surg 27 (7): 912-5, 1992.
- Szavay P, Luithle T, Semler O, et al.: Surgery of cavoatrial tumor thrombus in nephroblastoma: a report of the SIOP/GPOH study. Pediatr Blood Cancer 43 (1): 40-5, 2004.
- Powis M, Messahel B, Hobson R, et al.: Surgical complications after immediate nephrectomy versus preoperative chemotherapy in non-metastatic Wilms' tumour: findings from the 1991-2001 United Kingdom Children's Cancer Study Group UKW3 Trial. J Pediatr Surg 48 (11): 2181-6, 2013.
- Boam TD, Gabriel M, Shukla R, et al.: Impact of neoadjuvant chemotherapy on thrombus viability in patients with Wilms tumour and caval extension: systematic review with meta-analysis. BJS Open 5 (3): , 2021.
- Rutigliano DN, Kayton ML, Steinherz P, et al.: The use of preoperative chemotherapy in Wilms' tumor with contained retroperitoneal rupture. J Pediatr Surg 42 (9): 1595-9, 2007.
- Brisse HJ, Schleiermacher G, Sarnacki S, et al.: Preoperative Wilms tumor rupture: a retrospective study of 57 patients. Cancer 113 (1): 202-13, 2008.
- Corn BW, Goldwein JW, Evans I, et al.: Outcomes in low-risk babies treated with half-dose chemotherapy according to the Third National Wilms' Tumor Study. J Clin Oncol 10 (8): 1305-9, 1992.
- Morgan E, Baum E, Breslow N, et al.: Chemotherapy-related toxicity in infants treated according to the Second National Wilms' Tumor Study. J Clin Oncol 6 (1): 51-5, 1988.
- Green DM, Norkool P, Breslow NE, et al.: Severe hepatic toxicity after treatment with vincristine and dactinomycin using single-dose or divided-dose schedules: a report from the National Wilms' Tumor Study. J Clin Oncol 8 (9): 1525-30, 1990.
- Raine J, Bowman A, Wallendszus K, et al.: Hepatopathy-thrombocytopenia syndrome--a complication of dactinomycin therapy for Wilms' tumor: a report from the United Kingdom Childrens Cancer Study Group. J Clin Oncol 9 (2): 268-73, 1991.
- Oosterom N, Gooskens SLM, Renfro LA, et al.: Severe Hepatopathy in National Wilms Tumor Studies 3-5: Prevalence, Clinical Features, and Outcomes After Reintroduction of Chemotherapy. J Clin Oncol 41 (26): 4247-4256, 2023.
- Feusner JH, Ritchey ML, Norkool PA, et al.: Renal failure does not preclude cure in children receiving chemotherapy for Wilms tumor: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 242-5, 2008.
- Veal GJ, English MW, Grundy RG, et al.: Pharmacokinetically guided dosing of carboplatin in paediatric cancer patients with bilateral nephrectomy. Cancer Chemother Pharmacol 54 (4): 295-300, 2004.
- Dix DB, Fernandez CV, Chi YY, et al.: Augmentation of Therapy for Combined Loss of Heterozygosity 1p and 16q in Favorable Histology Wilms Tumor: A Children's Oncology Group AREN0532 and AREN0533 Study Report. J Clin Oncol 37 (30): 2769-2777, 2019.
- Stokes CL, Stokes WA, Kalapurakal JA, et al.: Timing of Radiation Therapy in Pediatric Wilms Tumor: A Report From the National Cancer Database. Int J Radiat Oncol Biol Phys 101 (2): 453-461, 2018.
- Dix DB, Seibel NL, Chi YY, et al.: Treatment of Stage IV Favorable Histology Wilms Tumor With Lung Metastases: A Report From the Children's Oncology Group AREN0533 Study. J Clin Oncol 36 (16): 1564-1570, 2018.
- Daw NC, Chi YY, Kalapurakal JA, et al.: Activity of Vincristine and Irinotecan in Diffuse Anaplastic Wilms Tumor and Therapy Outcomes of Stage II to IV Disease: Results of the Children's Oncology Group AREN0321 Study. J Clin Oncol 38 (14): 1558-1568, 2020.
- Thomas PR, Tefft M, Compaan PJ, et al.: Results of two radiation therapy randomizations in the third National Wilms' Tumor Study. Cancer 68 (8): 1703-7, 1991.
- Tefft M, D'Angio GJ, Beckwith B, et al.: Patterns of intra-abdominal relapse (IAR) in patients with Wilms' tumor who received radiation: analysis by histopathology. A report of National Wilms' Tumor Studies 1 and 2 (NWTS-1 & 2). Int J Radiat Oncol Biol Phys 6 (6): 663-7, 1980.
- Thomas PR, Tefft M, Farewell VT, et al.: Abdominal relapses in irradiated second National Wilms' Tumor Study patients. J Clin Oncol 2 (10): 1098-101, 1984.
- Meisel JA, Guthrie KA, Breslow NE, et al.: Significance and management of computed tomography detected pulmonary nodules: a report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 44 (3): 579-85, 1999.
- Dávila Fajardo R, Furtwängler R, van Grotel M, et al.: Outcome of Stage IV Completely Necrotic Wilms Tumour and Local Stage III Treated According to the SIOP 2001 Protocol. Cancers (Basel) 13 (5): , 2021.
- Vujanić GM, Graf N, D'Hooghe E, et al.: Omission of adjuvant chemotherapy in patients with completely necrotic Wilms tumor stage I and radiotherapy in stage III: The 30-year SIOP-RTSG experience. Pediatr Blood Cancer 71 (3): e30852, 2024.
- Daw NC, Chi YY, Kim Y, et al.: Treatment of stage I anaplastic Wilms' tumour: a report from the Children's Oncology Group AREN0321 study. Eur J Cancer 118: 58-66, 2019.
- Green DM, Breslow NE, Beckwith JB, et al.: Treatment with nephrectomy only for small, stage I/favorable histology Wilms' tumor: a report from the National Wilms' Tumor Study Group. J Clin Oncol 19 (17): 3719-24, 2001.
- Kalapurakal JA, Li SM, Breslow NE, et al.: Intraoperative spillage of favorable histology wilms tumor cells: influence of irradiation and chemotherapy regimens on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 76 (1): 201-6, 2010.
- Evageliou N, Renfro LA, Geller J, et al.: Prognostic impact of lymph node involvement and loss of heterozygosity of 1p or 16q in stage III favorable histology Wilms tumor: A report from Children's Oncology Group Studies AREN03B2 and AREN0532. Cancer 130 (5): 792-802, 2024.
- Benedetti DJ, Varela CR, Renfro LA, et al.: Treatment of children with favorable histology Wilms tumor with extrapulmonary metastases: A report from the COG studies AREN0533 and AREN03B2 and NWTSG study NWTS-5. Cancer 130 (6): 947-961, 2024.
- Grundy PE, Green DM, Dirks AC, et al.: Clinical significance of pulmonary nodules detected by CT and Not CXR in patients treated for favorable histology Wilms tumor on national Wilms tumor studies-4 and -5: a report from the Children's Oncology Group. Pediatr Blood Cancer 59 (4): 631-5, 2012.
- Verschuur A, Van Tinteren H, Graf N, et al.: Treatment of pulmonary metastases in children with stage IV nephroblastoma with risk-based use of pulmonary radiotherapy. J Clin Oncol 30 (28): 3533-9, 2012.
- Varan A, Büyükpamukçu N, Cağlar M, et al.: Prognostic significance of metastatic site at diagnosis in Wilms' tumor: results from a single center. J Pediatr Hematol Oncol 27 (4): 188-91, 2005.
- Szavay P, Luithle T, Graf N, et al.: Primary hepatic metastases in nephroblastoma--a report of the SIOP/GPOH Study. J Pediatr Surg 41 (1): 168-72; discussion 168-72, 2006.
- Fuchs J, Szavay P, Luithle T, et al.: Surgical implications for liver metastases in nephroblastoma--data from the SIOP/GPOH study. Surg Oncol 17 (1): 33-40, 2008.
- Aronson DC, Maharaj A, Sheik-Gafoor MH, et al.: The results of treatment of children with metastatic Wilms tumours (WT) in an African setting: do liver metastases have a negative impact on survival? Pediatr Blood Cancer 59 (2): 391-4, 2012.
- Liné A, Sudour-Bonnange H, Languillat-Fouquet V, et al.: Liver metastasis at diagnosis in children with nephroblastoma enrolled in SIOP2001 protocol: A French multicentric study. Pediatr Blood Cancer 67 (6): e28201, 2020.
- Kalapurakal JA, Pokhrel D, Gopalakrishnan M, et al.: Advantages of whole-liver intensity modulated radiation therapy in children with Wilms tumor and liver metastasis. Int J Radiat Oncol Biol Phys 85 (3): 754-60, 2013.
- Breslow NE, Collins AJ, Ritchey ML, et al.: End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol 174 (5): 1972-5, 2005.
- Aronson DC, Slaar A, Heinen RC, et al.: Long-term outcome of bilateral Wilms tumors (BWT). Pediatr Blood Cancer 56 (7): 1110-3, 2011.
- Zuppan CW, Beckwith JB, Weeks DA, et al.: The effect of preoperative therapy on the histologic features of Wilms' tumor. An analysis of cases from the Third National Wilms' Tumor Study. Cancer 68 (2): 385-94, 1991.
- Ehrlich PF: Bilateral Wilms' tumor: the need to improve outcomes. Expert Rev Anticancer Ther 9 (7): 963-73, 2009.
- Chintagumpala MM, Perlman EJ, Tornwall B, et al.: Outcomes based on histopathologic response to preoperative chemotherapy in children with bilateral Wilms tumor: A prospective study (COG AREN0534). Cancer 128 (13): 2493-2503, 2022.
- Romao RLP, Aldrink JH, Renfro LA, et al.: Bilateral Wilms tumor with anaplasia: A report from the Children's Oncology Group Study AREN0534. Pediatr Blood Cancer 71 (7): e30981, 2024.
- Sudour H, Audry G, Schleimacher G, et al.: Bilateral Wilms tumors (WT) treated with the SIOP 93 protocol in France: epidemiological survey and patient outcome. Pediatr Blood Cancer 59 (1): 57-61, 2012.
- Davidoff AM, Interiano RB, Wynn L, et al.: Overall Survival and Renal Function of Patients With Synchronous Bilateral Wilms Tumor Undergoing Surgery at a Single Institution. Ann Surg 262 (4): 570-6, 2015.
- Kieran K, Williams MA, McGregor LM, et al.: Repeat nephron-sparing surgery for children with bilateral Wilms tumor. J Pediatr Surg 49 (1): 149-53, 2014.
- Kist-van Holthe JE, Ho PL, Stablein D, et al.: Outcome of renal transplantation for Wilms' tumor and Denys-Drash syndrome: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Transplant 9 (3): 305-10, 2005.
- Venkatramani R, Chi YY, Coppes MJ, et al.: Outcome of patients with intracranial relapse enrolled on national Wilms Tumor Study Group clinical trials. Pediatr Blood Cancer 64 (7): , 2017.
- Iaboni DSM, Chi YY, Kim Y, et al.: Outcome of Wilms tumor patients with bone metastasis enrolled on National Wilms Tumor Studies 1-5: A report from the Children's Oncology Group. Pediatr Blood Cancer 66 (1): e27430, 2019.
- Malogolowkin M, Cotton CA, Green DM, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 50 (2): 236-41, 2008.
- Reinhard H, Schmidt A, Furtwängler R, et al.: Outcome of relapses of nephroblastoma in patients registered in the SIOP/GPOH trials and studies. Oncol Rep 20 (2): 463-7, 2008.
- Malogolowkin M, Spreafico F, Dome JS, et al.: Incidence and outcomes of patients with late recurrence of Wilms' tumor. Pediatr Blood Cancer 60 (10): 1612-5, 2013.
- Radhakrishnan V, Mishra S, Raja A, et al.: Relapse of Wilms tumor after 20 years: a rare presentation and review of literature. Pediatric Hematology Oncology Journal 1 (4): 86-8, 2016. Also available online. Last accessed December 22, 2022.
- Grundy P, Breslow N, Green DM, et al.: Prognostic factors for children with recurrent Wilms' tumor: results from the Second and Third National Wilms' Tumor Study. J Clin Oncol 7 (5): 638-47, 1989.
- Green DM, Cotton CA, Malogolowkin M, et al.: Treatment of Wilms tumor relapsing after initial treatment with vincristine and actinomycin D: a report from the National Wilms Tumor Study Group. Pediatr Blood Cancer 48 (5): 493-9, 2007.
- Warmann SW, Furtwängler R, Blumenstock G, et al.: Tumor biology influences the prognosis of nephroblastoma patients with primary pulmonary metastases: results from SIOP 93-01/GPOH and SIOP 2001/GPOH. Ann Surg 254 (1): 155-62, 2011.
- Groenendijk A, van Tinteren H, Jiang Y, et al.: Outcome of SIOP patients with low- or intermediate-risk Wilms tumour relapsing after initial vincristine and actinomycin-D therapy only - the SIOP 93-01 and 2001 protocols. Eur J Cancer 163: 88-97, 2022.
- Abu-Ghosh AM, Krailo MD, Goldman SC, et al.: Ifosfamide, carboplatin and etoposide in children with poor-risk relapsed Wilms' tumor: a Children's Cancer Group report. Ann Oncol 13 (3): 460-9, 2002.
- Garaventa A, Hartmann O, Bernard JL, et al.: Autologous bone marrow transplantation for pediatric Wilms' tumor: the experience of the European Bone Marrow Transplantation Solid Tumor Registry. Med Pediatr Oncol 22 (1): 11-4, 1994.
- Pein F, Michon J, Valteau-Couanet D, et al.: High-dose melphalan, etoposide, and carboplatin followed by autologous stem-cell rescue in pediatric high-risk recurrent Wilms' tumor: a French Society of Pediatric Oncology study. J Clin Oncol 16 (10): 3295-301, 1998.
- Rossoff J, Tse WT, Duerst RE, et al.: High-dose chemotherapy and autologous hematopoietic stem-cell rescue for treatment of relapsed and refractory Wilms tumor: Re-evaluating outcomes. Pediatr Hematol Oncol 35 (5-6): 316-321, 2018 Aug - Sep.
- Delafoy M, Verschuur A, Scheleirmacher G, et al.: High-dose chemotherapy followed by autologous stem cell rescue in Wilms tumors: French report on toxicity and efficacy. Pediatr Blood Cancer 69 (3): e29431, 2022.
- Spreafico F, Dalissier A, Pötschger U, et al.: High dose chemotherapy and autologous hematopoietic cell transplantation for Wilms tumor: a study of the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant 55 (2): 376-383, 2020.
- Malogolowkin MH, Hemmer MT, Le-Rademacher J, et al.: Outcomes following autologous hematopoietic stem cell transplant for patients with relapsed Wilms' tumor: a CIBMTR retrospective analysis. Bone Marrow Transplant 52 (11): 1549-1555, 2017.
- Weil BR, Murphy AJ, Liu Q, et al.: Late Health Outcomes Among Survivors of Wilms Tumor Diagnosed Over Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol 41 (14): 2638-2650, 2023.
- Wong KF, Reulen RC, Winter DL, et al.: Risk of Adverse Health and Social Outcomes Up to 50 Years After Wilms Tumor: The British Childhood Cancer Survivor Study. J Clin Oncol 34 (15): 1772-9, 2016.
- Foster KL, Salehabadi SM, Green DM, et al.: Clinical Assessment of Late Health Outcomes in Survivors of Wilms Tumor. Pediatrics 150 (5): , 2022.
- Lange JM, Takashima JR, Peterson SM, et al.: Breast cancer in female survivors of Wilms tumor: a report from the national Wilms tumor late effects study. Cancer 120 (23): 3722-30, 2014.
- Green DM, Grigoriev YA, Nan B, et al.: Congestive heart failure after treatment for Wilms' tumor: a report from the National Wilms' Tumor Study group. J Clin Oncol 19 (7): 1926-34, 2001.
- Practice Committee of the American Society for Reproductive Medicine. Electronic address: [email protected]: Fertility preservation in patients undergoing gonadotoxic therapy or gonadectomy: a committee opinion. Fertil Steril 112 (6): 1022-1033, 2019.
- Green DM, Liu W, Kutteh WH, et al.: Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: a report from the St Jude Lifetime Cohort Study. Lancet Oncol 15 (11): 1215-23, 2014.
- Green DM, Lange JM, Peabody EM, et al.: Pregnancy outcome after treatment for Wilms tumor: a report from the national Wilms tumor long-term follow-up study. J Clin Oncol 28 (17): 2824-30, 2010.
- Breslow NE, Takashima JR, Ritchey ML, et al.: Renal failure in the Denys-Drash and Wilms' tumor-aniridia syndromes. Cancer Res 60 (15): 4030-2, 2000.

