Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre el osteosarcoma y el sarcoma pleomórfico indiferenciado óseo (antes llamado histiocitoma fibroso maligno óseo, HFM)
Aspectos generales de la enfermedad
El osteosarcoma se presenta de forma predominante en adolescentes y adultos jóvenes. En una revisión de los datos del National Childhood Cancer Registry del Instituto Nacional del Cáncer se calculó una tasa de incidencia anual de osteosarcoma de 5,4 casos por millón en personas de 0 a 19 años y de 4 casos por millón en personas de menos de 40 años.[1] La Oficina del Censo de los Estados Unidos calculó que había 82 millones de personas entre los 0 y 19 años de edad, lo que se traduce en una incidencia de alrededor de 440 casos anuales en este grupo de edad.
El osteosarcoma representa cerca del 5 % de los tumores infantiles. En los niños y adolescentes, más del 50 % de estos tumores surgen en los huesos largos alrededor de la rodilla. El osteosarcoma en tejido blando o en vísceras es muy infrecuente. No hay ninguna diferencia en cuanto a los síntomas de presentación inicial, la ubicación del tumor y el desenlace en los pacientes más jóvenes (<12 años), en comparación con los adolescentes.[2,3]
En la década de 1980, se realizaron dos ensayos clínicos diseñados para determinar si la quimioterapia alteraba la evolución natural del osteosarcoma después de la extirpación quirúrgica del tumor primario. El desenlace de estos pacientes permitió recapitular la experiencia anterior a 1970: más de la mitad de estos pacientes presentaron metástasis en el transcurso de los 6 meses posteriores al diagnóstico y, en general, alrededor del 90 % presentó recidiva de la enfermedad en el transcurso de los 2 años posteriores al diagnóstico.[4] La supervivencia general (SG) de los pacientes tratados con cirugía sola fue estadísticamente inferior.[5] La evolución natural del osteosarcoma no ha cambiado con el tiempo; se anticipa que menos del 20 % de los pacientes con tumores primarios localizados y resecables tratados con cirugía sola sobrevivan sin recaída.[4,6]; [7][Nivel de evidencia A1]
En 2013, la Organización Mundial de la Salud (OMS) actualizó la clasificación de los tumores de tejido blando y hueso.[8] Se eliminó el término histiocitoma fibroso maligno (HFM) y se reemplazó por el de sarcoma pleomórfico indiferenciado (SPI). Aunque este tipo de sarcoma es mucho más frecuente en los tejidos blandos, también surge en los huesos, en donde exhibe características histológicas similares al osteosarcoma, pero no produce matriz osteoide. La mayor parte de la bibliografía que describe el comportamiento clínico y la respuesta al tratamiento de este tipo histológico óseo se publicó antes de la actualización de 2013 de la OMS, de manera que si se realiza una búsqueda con el término en inglés UPS of bone (sarcoma pleomórfico indiferenciado [SPI] óseo) no se identificarán estos artículos. Las referencias bibliográficas de este resumen aparecen con sus títulos tal como se publicaron; por lo tanto, en muchas referencias se usa el término MFH of bone (histiocitoma fibroso maligno óseo, HFM), afección que en la actualidad se llama UPS of bone.
Evaluación diagnóstica
El osteosarcoma se diagnostica mediante una biopsia con aguja gruesa o una biopsia quirúrgica abierta. Es preferible que la biopsia la realice un cirujano experto en técnicas de conservación del miembro (extirpación del tumor óseo maligno sin amputación ni reemplazo óseo o articular con aloinjertos o prótesis). En estos casos, la ubicación de la incisión de la biopsia original es crucial. Una alineación inapropiada de la biopsia o la contaminación inadvertida de los tejidos blandos quizás imposibilite la cirugía reconstructiva para conservar el miembro.
Factores pronósticos
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer. Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más de un 50 %.[1,9,10] Con respecto al osteosarcoma, la tasa de supervivencia relativa a 5 años aumentó durante el mismo período del 40 % al 72 % en niños menores de 15 años, y del 56 % a alrededor del 71 % en adolescentes de 15 a 19 años; no obstante, no se ha producido ninguna mejora importante desde la década de 1980.[1,11]
En general, los factores pronósticos del osteosarcoma no han sido útiles para identificar a los pacientes que se podrían beneficiar de la intensificación del tratamiento, o a aquellos que tal vez necesiten menos tratamiento, pero que mantengan un resultado excelente.
Los factores que afectan el desenlace son los siguientes:[12]
- Sitio del tumor primario.
- Tamaño del tumor primario.
- Presencia de enfermedad metastásica clínicamente detectable.
- Resecabilidad quirúrgica del tumor primario.
- Grado de necrosis tumoral después de la administración de quimioterapia neoadyuvante.
- Edad y sexo.
- Otros factores pronósticos posibles.
Sitio del tumor primario
El sitio del tumor primario es un factor pronóstico significativo en los pacientes con enfermedad localizada. Entre los tumores de las extremidades, los sitios distales tienen un pronóstico más favorable que los sitios proximales. Los tumores primarios del esqueleto axial se relacionan con el riesgo más alto de progresión y muerte, sobre todo porque no es posible lograr una resección quirúrgica completa.
Las consideraciones pronósticas para los sitios en el esqueleto axial y para los sitios extraesqueléticos son las siguientes:
- Pelvis. Los osteosarcomas pélvicos abarcan entre el 7 % y el 9 % de todos los osteosarcomas; las tasas de supervivencia para pacientes con tumores primarios en la pelvis oscilan entre el 20 % y el 47 %.[13,14,15] La resección quirúrgica completa se relaciona con un desenlace favorable para el osteosarcoma de la pelvis en algunas cohortes de pacientes.[13,16]
- Craneofacial, o de cabeza y cuello. En los pacientes con osteosarcoma craneofacial, aquellos con sitios primarios en el maxilar inferior y superior tienen mejor pronóstico que los pacientes con otros sitios primarios en la cabeza y el cuello.[17,18,19] Para los pacientes con osteosarcoma en los huesos craneofaciales, la resección completa del tumor primario con márgenes negativos es esencial para la curación.[20,21,22] Cuando se tratan con cirugía sola, los pacientes con osteosarcoma de cabeza y cuello tienen mejor pronóstico que aquellos con lesiones en las extremidades. Sin embargo, no se recomienda la cirugía sola sin terapia adyuvante para el osteosarcoma de cabeza y cuello de grado alto.
A pesar de la tasa relativamente alta de necrosis inferior después de la quimioterapia neoadyuvante, las metástasis sistémicas se presentan en menos pacientes con tumores primarios craneofaciales que en los pacientes con osteosarcomas que se originan en las extremidades. Es posible que esto se deba a la inclusión de pacientes con tumores de grado más bajo en las cohortes notificadas.[23,24,25]
En un metanálisis se concluyó que la quimioterapia sistémica adyuvante mejora el pronóstico de los pacientes con osteosarcoma de cabeza y cuello; aunque, en series pequeñas no se observó dicho beneficio.[23,24,25] En otro metanálisis grande no se encontró ningún beneficio de la quimioterapia para los pacientes con osteosarcoma de cabeza y cuello; sin embargo, se indicó que incluir la quimioterapia al tratamiento de los pacientes con tumores de grado alto quizás mejore la supervivencia.[22] En un análisis retrospectivo se identificó una tendencia hacia una mejor supervivencia en pacientes con osteosarcoma de grado alto en el maxilar inferior y superior que recibieron quimioterapia adyuvante.[22,26]
En un estudio retrospectivo se encontró que la radioterapia mejora el control local, la supervivencia específica de la enfermedad y la SG en los pacientes con osteosarcoma de los huesos craneofaciales que presentaron márgenes positivos o indeterminados después de la resección quirúrgica.[27][Nivel de evidencia C1] Los osteosarcomas craneofaciales relacionados con la radiación por lo general son lesiones de grado alto, a menudo fibroblásticas, y suelen recidivar de manera local con una tasa alta de metástasis.[28]
- Extraesquelético. El osteosarcoma en sitios extraesqueléticos es raro en niños y adultos jóvenes. La modalidad de terapia combinada actual en los pacientes con osteosarcoma extraesquelético produce desenlaces similares a los de los pacientes con tumores primarios de hueso.[29]
Tamaño del tumor primario
En algunas series, los pacientes con tumores más grandes presentaron un pronóstico más precario que los pacientes con tumores más pequeños.[12,30,31] El tamaño del tumor se determinó mediante la dimensión más larga, el área transversal o por estimación del volumen tumoral; todas las evaluaciones se correlacionaron con el desenlace.
Es probable que la concentración de lactato–deshidrogenasa (LDH) sérica elevada, que también se correlaciona con un desenlace más precario, sea un marcador indirecto del volumen tumoral.[14]
Presencia de enfermedad metastásica clínicamente detectable
Los pacientes con enfermedad localizada tienen un pronóstico mucho más favorable que los pacientes con enfermedad metastásica evidente. Hasta un 20 % de los pacientes tienen metástasis detectables mediante radiografías en el momento del diagnóstico, siendo los pulmones los sitios más comunes.[32] El pronóstico de los pacientes con enfermedad metastásica se determina en gran medida por el sitio y el número de las metástasis, así como el grado de resecabilidad quirúrgica de la enfermedad metastásica.[33,34]
- Sitio de las metástasis: el pronóstico es más favorable en los pacientes con menos nódulos pulmonares y para aquellos con metástasis pulmonares unilaterales en lugar de bilaterales.[33] No todos los pacientes con sospecha de metástasis pulmonares en el momento del diagnóstico presentan osteosarcoma confirmado en el momento de la resección pulmonar. En una serie grande, alrededor del 25 % de los pacientes solo presentaron lesiones benignas extirpadas en el momento de la cirugía.[34]
- Número de metástasis: se notificó que los pacientes con metástasis discontinuas (por lo menos 2 lesiones discontinuas en el mismo hueso) tienen un pronóstico inferior.[35] No obstante, en un análisis del German Cooperative Osteosarcoma Study Group (COSS) se indica que las lesiones discontinuas en el mismo hueso no confieren un pronóstico inferior si se incluyen en la resección quirúrgica planificada. Las metástasis discontinuas en un hueso distinto al del tumor primario se deben considerar metástasis sistémicas.[36]
Tradicionalmente, la metástasis en una articulación se definía como lesión discontinua, pero en una clasificación posterior del American Joint Committee on Cancer se excluyó a tales lesiones como lesiones discontinuas.[37] Es posible que se consideraran diseminación hemática y acarreen un pronóstico más adverso.[36]
Los pacientes con osteosarcoma multifocal (definido como lesiones óseas múltiples sin tumor primario claro) tienen un pronóstico muy precario.[38,39]
- Resecabilidad quirúrgica de las metástasis: los pacientes que se someten a una ablación quirúrgica completa del tumor primario y metastásico (cuando está confinado al pulmón) después de la quimioterapia a veces sobreviven a largo plazo, pero las tasas generales de supervivencia sin complicaciones (SSC) aún se sitúan en el intervalo del 20 % al 30 % para los pacientes con enfermedad metastásica en el momento del diagnóstico.[33,34,40,41] Los pacientes con osteosarcoma metastásico fueron aptos para participar en el European and American Osteosarcoma Study (EURAMOS) solo si tenían una enfermedad potencialmente resecable. Aunque los pacientes con enfermedad metastásica tuvieron una tasa general de SSC a 5 años de solo el 28 %, aquellos que lograron una remisión quirúrgica completa en todos los sitios (3–6 meses después del diagnóstico) tuvieron una tasa de SSC a 5 años del 64 % y una tasa de SG del 79 %.[31]
Resecabilidad quirúrgica del tumor primario
La resecabilidad del tumor es una característica pronóstica muy importante y, por lo general, la resección completa del tumor primario y de cualquier lesión discontinua con márgenes adecuados se considera esencial para la curación. Es posible que la radioterapia mejore la supervivencia de los pacientes con tumores primarios del esqueleto axial que no se someten a cirugía del tumor primario, o que se someten a una cirugía en la que se obtienen márgenes con compromiso tumoral.[16,42]
En una revisión retrospectiva de pacientes con osteosarcoma craneofacial, dirigida por el Cooperative German-Austrian-Swiss Osteosarcoma Study Group, se notificó que la resección quirúrgica incompleta se relacionó con una probabilidad de supervivencia más baja.[17][Nivel de evidencia C1] En un estudio cooperativo realizado en Europa, el tamaño del margen no fue significativo. No obstante, el pronóstico fue mejor cuando la biopsia y la resección se llevaron a cabo en un centro con experiencia en oncología ortopédica.[14]
Grado de necrosis tumoral después de quimioterapia neoadyuvante
En la mayoría de los protocolos de tratamiento del osteosarcoma, se utiliza un período inicial de quimioterapia sistémica antes de la resección definitiva del tumor primario (o la resección de los sitios con metástasis). El patólogo evalúa la necrosis en el tumor resecado. Los pacientes con un mínimo del 90 % de necrosis en el tumor primario después de la quimioterapia de inducción tienen un mejor pronóstico que quienes presentan menos necrosis.[30] Los pacientes con menos necrosis (<90 %) en el tumor primario después de la quimioterapia inicial tienen una tasa más alta de recidiva en los primeros 2 años que los pacientes con una cantidad más favorable de necrosis (≥90 %).[43]
Una necrosis más baja no se debe interpretar como ineficacia quimioterapéutica; las tasas de curación en los pacientes con poca o ninguna necrosis luego de la quimioterapia de inducción son mucho más altas que las tasas de curación en los pacientes que no reciben quimioterapia. La tasa de SSC para los pacientes que no reciben quimioterapia adyuvante es de cerca del 11 %.[44] En muchas series grandes publicadas de pacientes tratados con quimioterapia se notificaron tasas de SSC del 40 % al 50 % en los pacientes con poca o ninguna necrosis en el tumor primario después de la quimioterapia sistémica inicial.[45,46,47] En una revisión de dos ensayos prospectivos consecutivos, realizados por el Children's Oncology Group, se observó que la duración y la intensidad del período inicial de quimioterapia afectaron la necrosis histológica del tumor primario después de la quimioterapia inicial. Cuanto mayor fue la necrosis, mejor fue el desenlace en ambos ensayos; sin embargo, la magnitud de la diferencia entre los pacientes con más o menos necrosis disminuyó a medida que se prologaba e intensificaba el período de la quimioterapia inicial.[48][Nivel de evidencia B1]
Edad y sexo
Los pacientes del grupo etario de adolescentes mayores y adultos jóvenes que, por lo general, se define por edades entre 18 y 40 años, suelen presentar un pronóstico más precario. Además, el sexo masculino se ha vinculado con un pronóstico más precario.[31,49,50] En comparación con otros factores pronósticos enumerados, tanto la edad como el sexo tienen un efecto relativamente menor en el desenlace.
Efecto del paso del tiempo sobre los factores pronósticos
En un análisis del EURAMOS se encontró que el poder predictivo de los factores pronósticos iniciales cambia con el tiempo.[51] Se aplicó un enfoque de referencia a 1965 pacientes. Los resultados demostraron que la recidiva local y las metástasis nuevas afectaron de manera negativa la SG a 5 años (cociente de riesgos instantáneos [CRI] de la recidiva local, 2,634; IC 95 %, 1,845–3,761; y CRI de metástasis, 8,558; IC 95 %, 7,367–9,942). Los factores iniciales con valor pronóstico negativo sólido (CRI >2) fueron: respuesta histológica precaria (≥10 % tumor viable), ubicación axial del tumor y presencia de metástasis pulmonar. El efecto de una buena respuesta histológica versus una respuesta histológica precaria cambió con el tiempo y dejó de ser significativo a partir de 3,25 años desde la cirugía.
El COSS alemán analizó su base de datos de 5503 pacientes con osteosarcoma registrados entre 1980 y 2019. Se identificó a 2009 pacientes que habían sobrevivido más de 5 años desde la biopsia diagnóstica para evaluar los factores pronósticos de la supervivencia a largo plazo.[52] Los autores confirmaron los factores pronósticos conocidos de fracaso del tratamiento, como una menor necrosis después de la quimioterapia inicial, mayor edad en el momento del diagnóstico, tumor en un sitio desfavorable y presencia de una segunda neoplasia maligna. Se notificaron las tasas de SG pasados los 5 años (consultar el Cuadro 1).
| COSS = Cooperative Osteosarcoma Study Group; SG = supervivencia general. | ||||
| Seguimiento adicional | 5 años | 10 años | 15 años | 20 años |
| Número de pacientes analizados en cada intervalo de tiempo | 161 | 808 | 288 | 125 |
| Tasa de SG | 91,7 % | 88,9 % | 85,8 % | 83,4 % |
En un análisis multivariante, los factores que conservaron importancia para una supervivencia a largo plazo más precaria fueron la aparición de una recidiva en el periodo de 1 a 5 años, mayor edad en el momento del diagnóstico y presencia de una segunda neoplasia maligna. La extensión de la necrosis tumoral después de la quimioterapia inicial no tuvo validez pasados los 5 años de seguimiento.[52]
Otros factores pronósticos posibles
Otros factores que a veces son pronósticos, pero sobre los que se dispone de datos limitados o contradictorios, son los siguientes:
- Neoplasias subsiguientes. Los pacientes que tienen un osteosarcoma clasificado como neoplasia subsiguiente, como los tumores que surgen en un campo de radiación, comparten el mismo pronóstico de los pacientes con un osteosarcoma nuevo cuando reciben tratamiento intensivo con resección quirúrgica completa y quimioterapia multifarmacológica.[53,54]
En una serie alemana, de los pacientes con osteosarcoma craneofacial, cerca del 25 % presentaba un osteosarcoma como segundo tumor y, en 8 de estos 13 casos, el osteosarcoma apareció después del tratamiento de un retinoblastoma. En esta serie, no hubo diferencia en el desenlace del osteosarcoma craneofacial primario o secundario.[17]
- Anomalías en las pruebas de laboratorio. Los posibles factores pronósticos que se identificaron en los pacientes con osteosarcoma clásico de grado alto localizado son las concentraciones de LDH y fosfatasa alcalina, y el subtipo histológico.[30,46,49,55,56,57,58]
- Índice de masa corporal. Un índice de masa corporal más alto en el momento de la presentación inicial se relaciona con una SG más precaria.[59]
- Fractura patológica. En algunos estudios, se indicó que una fractura patológica en el momento del diagnóstico o durante la quimioterapia preoperatoria no tiene un significado de pronóstico adverso.[6]; [60,61][Nivel de evidencia C1]; [62][Nivel de evidencia C2]
Sin embargo, en una revisión sistemática de 9 estudios de cohorte se evaluó el efecto de las fracturas patológicas en el desenlace de pacientes con osteosarcoma. La revisión incluyó a 2187 pacientes, 311 de ellos con fractura patológica. La presencia de fractura patológica se correlacionó con disminuciones en la SSC y la SG.[63] En otras dos series, una fractura patológica en el momento del diagnóstico se relacionó con un desenlace general más precario.[64]; [65][Nivel de evidencia C1] En un análisis retrospectivo de 2847 pacientes con osteosarcoma del COSS alemán, se identificaron 321 pacientes (11,3 %) con fractura patológica antes del comienzo del tratamiento sistémico.[66][Nivel de evidencia C1] En los pacientes pediátricos, la SG y la SSC no difirieron significativamente entre los pacientes con fractura patológica o sin esta. La tasa de SG a 5 años fue del 46 % en adultos con una fractura patológica versus el 69 % en adultos sin una fractura patológica (P < 0,001). La tasa de SSC a 5 años fue del 36 % en adultos con fractura patológica versus el 56 % en adultos sin fractura patológica (P < 0,001). En un análisis multivariante, la presencia de fractura patológica no fue un factor estadísticamente significativo para la SG o la SSC en la cohorte total ni en los pacientes pediátricos. En adultos, la presencia de una fractura patológica permaneció como un factor pronóstico independiente para la SG (CRI, 1,893; P = 0,013).
- Tiempo transcurrido hasta la cirugía definitiva. En una serie grande, se encontró que una demora de 21 días o más desde el momento de la cirugía definitiva hasta la reanudación de la quimioterapia fue un factor de pronóstico adverso.[67]
- Factores genéticos.
- Expresión de ERBB2. Hay datos contradictorios sobre la importancia pronóstica de este factor de crecimiento epidérmico humano.[68,69,70]
- Ploidia de células tumorales.[71]
- Pérdidas y ganancias cromosómicas específicas.[72]
- Pérdida de heterocigosidad del gen RB1.[73,74]
- Pérdida de heterocigosidad del locus TP53.[75]
- Aumento de expresión de la glicoproteína p.[76,77] En un análisis prospectivo de la expresión de la glicoproteína p determinada por inmunohistoquímica, no se logró definir su importancia pronóstica en pacientes con diagnóstico reciente de osteosarcoma; sin embargo, en estudios previos se indicó que la sobreexpresión de la glicoproteína p predijo un desenlace precario.[78]
Para obtener más información, consultar la sección Características genómicas del osteosarcoma.
Referencias:
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
- Bacci G, Longhi A, Bertoni F, et al.: Primary high-grade osteosarcoma: comparison between preadolescent and older patients. J Pediatr Hematol Oncol 27 (3): 129-34, 2005.
- Bacci G, Balladelli A, Palmerini E, et al.: Neoadjuvant chemotherapy for osteosarcoma of the extremities in preadolescent patients: the Rizzoli Institute experience. J Pediatr Hematol Oncol 30 (12): 908-12, 2008.
- Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
- Link MP: The multi-institutional osteosarcoma study: an update. Cancer Treat Res 62: 261-7, 1993.
- Bacci G, Ferrari S, Longhi A, et al.: Nonmetastatic osteosarcoma of the extremity with pathologic fracture at presentation: local and systemic control by amputation or limb salvage after preoperative chemotherapy. Acta Orthop Scand 74 (4): 449-54, 2003.
- Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
- Fletcher CDM, Bridge JA, Hogendoorn P, et al., eds.: WHO Classification of Tumours of Soft Tissue and Bone. 4th ed. IARC Press, 2013.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
- Isakoff MS, Bielack SS, Meltzer P, et al.: Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol 33 (27): 3029-35, 2015.
- Pakos EE, Nearchou AD, Grimer RJ, et al.: Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer 45 (13): 2367-75, 2009.
- Donati D, Giacomini S, Gozzi E, et al.: Osteosarcoma of the pelvis. Eur J Surg Oncol 30 (3): 332-40, 2004.
- Andreou D, Bielack SS, Carrle D, et al.: The influence of tumor- and treatment-related factors on the development of local recurrence in osteosarcoma after adequate surgery. An analysis of 1355 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Ann Oncol 22 (5): 1228-35, 2011.
- Isakoff MS, Barkauskas DA, Ebb D, et al.: Poor survival for osteosarcoma of the pelvis: a report from the Children's Oncology Group. Clin Orthop Relat Res 470 (7): 2007-13, 2012.
- Ozaki T, Flege S, Kevric M, et al.: Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 21 (2): 334-41, 2003.
- Jasnau S, Meyer U, Potratz J, et al.: Craniofacial osteosarcoma Experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol 44 (3): 286-94, 2008.
- Laskar S, Basu A, Muckaden MA, et al.: Osteosarcoma of the head and neck region: lessons learned from a single-institution experience of 50 patients. Head Neck 30 (8): 1020-6, 2008.
- Kassir RR, Rassekh CH, Kinsella JB, et al.: Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope 107 (1): 56-61, 1997.
- Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
- Smith RB, Apostolakis LW, Karnell LH, et al.: National Cancer Data Base report on osteosarcoma of the head and neck. Cancer 98 (8): 1670-80, 2003.
- Fernandes R, Nikitakis NG, Pazoki A, et al.: Osteogenic sarcoma of the jaw: a 10-year experience. J Oral Maxillofac Surg 65 (7): 1286-91, 2007.
- Smeele LE, Kostense PJ, van der Waal I, et al.: Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol 15 (1): 363-7, 1997.
- Ha PK, Eisele DW, Frassica FJ, et al.: Osteosarcoma of the head and neck: a review of the Johns Hopkins experience. Laryngoscope 109 (6): 964-9, 1999.
- Duffaud F, Digue L, Baciuchka-Palmaro M, et al.: Osteosarcomas of flat bones in adolescents and adults. Cancer 88 (2): 324-32, 2000.
- Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group: Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol 33 (3): 139-44, 2004.
- Guadagnolo BA, Zagars GK, Raymond AK, et al.: Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer 115 (14): 3262-70, 2009.
- McHugh JB, Thomas DG, Herman JM, et al.: Primary versus radiation-associated craniofacial osteosarcoma: Biologic and clinicopathologic comparisons. Cancer 107 (3): 554-62, 2006.
- Goldstein-Jackson SY, Gosheger G, Delling G, et al.: Extraskeletal osteosarcoma has a favourable prognosis when treated like conventional osteosarcoma. J Cancer Res Clin Oncol 131 (8): 520-6, 2005.
- Bielack SS, Kempf-Bielack B, Delling G, et al.: Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. J Clin Oncol 20 (3): 776-90, 2002.
- Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
- Meyers PA, Schwartz CL, Krailo M, et al.: Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 23 (9): 2004-11, 2005.
- Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
- Bacci G, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. J Surg Oncol 98 (6): 415-20, 2008.
- Sajadi KR, Heck RK, Neel MD, et al.: The incidence and prognosis of osteosarcoma skip metastases. Clin Orthop Relat Res (426): 92-6, 2004.
- Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
- American Joint Committee on Cancer: AJCC Cancer Staging Manual. 6th ed. Springer, 2002.
- Bacci G, Fabbri N, Balladelli A, et al.: Treatment and prognosis for synchronous multifocal osteosarcoma in 42 patients. J Bone Joint Surg Br 88 (8): 1071-5, 2006.
- Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
- Goorin AM, Shuster JJ, Baker A, et al.: Changing pattern of pulmonary metastases with adjuvant chemotherapy in patients with osteosarcoma: results from the multiinstitutional osteosarcoma study. J Clin Oncol 9 (4): 600-5, 1991.
- Bacci G, Mercuri M, Longhi A, et al.: Grade of chemotherapy-induced necrosis as a predictor of local and systemic control in 881 patients with non-metastatic osteosarcoma of the extremities treated with neoadjuvant chemotherapy in a single institution. Eur J Cancer 41 (14): 2079-85, 2005.
- DeLaney TF, Park L, Goldberg SI, et al.: Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys 61 (2): 492-8, 2005.
- Kim MS, Cho WH, Song WS, et al.: time dependency of prognostic factors in patients with stage II osteosarcomas. Clin Orthop Relat Res 463: 157-65, 2007.
- Link MP, Goorin AM, Horowitz M, et al.: Adjuvant chemotherapy of high-grade osteosarcoma of the extremity. Updated results of the Multi-Institutional Osteosarcoma Study. Clin Orthop (270): 8-14, 1991.
- Winkler K, Beron G, Delling G, et al.: Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J Clin Oncol 6 (2): 329-37, 1988.
- Meyers PA, Heller G, Healey J, et al.: Chemotherapy for nonmetastatic osteogenic sarcoma: the Memorial Sloan-Kettering experience. J Clin Oncol 10 (1): 5-15, 1992.
- Bielack S, Jürgens H, Jundt G, et al.: Osteosarcoma: the COSS experience. Cancer Treat Res 152: 289-308, 2009.
- Bishop MW, Chang YC, Krailo MD, et al.: Assessing the Prognostic Significance of Histologic Response in Osteosarcoma: A Comparison of Outcomes on CCG-782 and INT0133-A Report From the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 63 (10): 1737-43, 2016.
- Janeway KA, Barkauskas DA, Krailo MD, et al.: Outcome for adolescent and young adult patients with osteosarcoma: a report from the Children's Oncology Group. Cancer 118 (18): 4597-605, 2012.
- Collins M, Wilhelm M, Conyers R, et al.: Benefits and adverse events in younger versus older patients receiving neoadjuvant chemotherapy for osteosarcoma: findings from a meta-analysis. J Clin Oncol 31 (18): 2303-12, 2013.
- Spreafico M, Hazewinkel AD, Gelderblom H, et al.: Dynamic Prediction of Overall Survival for Patients with Osteosarcoma: A Retrospective Analysis of the EURAMOS-1 Clinical Trial Data. Curr Oncol 31 (7): 3630-3642, 2024.
- Fernandes JS, Blattmann C, Hecker-Nolting S, et al.: Beyond 5-year survival. A report from the Cooperative Osteosarcoma Study Group (COSS). Cancer Med 13 (1): e6893, 2024.
- Tabone MD, Terrier P, Pacquement H, et al.: Outcome of radiation-related osteosarcoma after treatment of childhood and adolescent cancer: a study of 23 cases. J Clin Oncol 17 (9): 2789-95, 1999.
- Bielack SS, Blattmann C, Hassenpflug W, et al.: Osteosarcoma Arising After Ewing Sarcoma or Vice Versa: A Report of 20 Affected Patients from the Cooperative Osteosarcoma Study Group (COSS). Anticancer Res 43 (11): 4975-4981, 2023.
- Bacci G, Longhi A, Versari M, et al.: Prognostic factors for osteosarcoma of the extremity treated with neoadjuvant chemotherapy: 15-year experience in 789 patients treated at a single institution. Cancer 106 (5): 1154-61, 2006.
- Bieling P, Rehan N, Winkler P, et al.: Tumor size and prognosis in aggressively treated osteosarcoma. J Clin Oncol 14 (3): 848-58, 1996.
- Ferrari S, Bertoni F, Mercuri M, et al.: Predictive factors of disease-free survival for non-metastatic osteosarcoma of the extremity: an analysis of 300 patients treated at the Rizzoli Institute. Ann Oncol 12 (8): 1145-50, 2001.
- Kager L, Zoubek A, Dominkus M, et al.: Osteosarcoma in very young children: experience of the Cooperative Osteosarcoma Study Group. Cancer 116 (22): 5316-24, 2010.
- Altaf S, Enders F, Jeavons E, et al.: High-BMI at diagnosis is associated with inferior survival in patients with osteosarcoma: a report from the Children's Oncology Group. Pediatr Blood Cancer 60 (12): 2042-6, 2013.
- Kim MS, Lee SY, Lee TR, et al.: Prognostic effect of pathologic fracture in localized osteosarcoma: a cohort/case controlled study at a single institute. J Surg Oncol 100 (3): 233-9, 2009.
- Xie L, Guo W, Li Y, et al.: Pathologic fracture does not influence local recurrence and survival in high-grade extremity osteosarcoma with adequate surgical margins. J Surg Oncol 106 (7): 820-5, 2012.
- Chung LH, Wu PK, Chen CF, et al.: Pathological fractures in predicting clinical outcomes for patients with osteosarcoma. BMC Musculoskelet Disord 17 (1): 503, 2016.
- Sun L, Li Y, Zhang J, et al.: Prognostic value of pathologic fracture in patients with high grade localized osteosarcoma: a systemic review and meta-analysis of cohort studies. J Orthop Res 33 (1): 131-9, 2015.
- Bramer JA, Abudu AA, Grimer RJ, et al.: Do pathological fractures influence survival and local recurrence rate in bony sarcomas? Eur J Cancer 43 (13): 1944-51, 2007.
- Schlegel M, Zeumer M, Prodinger PM, et al.: Impact of Pathological Fractures on the Prognosis of Primary Malignant Bone Sarcoma in Children and Adults: A Single-Center Retrospective Study of 205 Patients. Oncology 94 (6): 354-362, 2018.
- Kelley LM, Schlegel M, Hecker-Nolting S, et al.: Pathological Fracture and Prognosis of High-Grade Osteosarcoma of the Extremities: An Analysis of 2,847 Consecutive Cooperative Osteosarcoma Study Group (COSS) Patients. J Clin Oncol 38 (8): 823-833, 2020.
- Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
- Gorlick R, Huvos AG, Heller G, et al.: Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol 17 (9): 2781-8, 1999.
- Onda M, Matsuda S, Higaki S, et al.: ErbB-2 expression is correlated with poor prognosis for patients with osteosarcoma. Cancer 77 (1): 71-8, 1996.
- Kilpatrick SE, Geisinger KR, King TS, et al.: Clinicopathologic analysis of HER-2/neu immunoexpression among various histologic subtypes and grades of osteosarcoma. Mod Pathol 14 (12): 1277-83, 2001.
- Look AT, Douglass EC, Meyer WH: Clinical importance of near-diploid tumor stem lines in patients with osteosarcoma of an extremity. N Engl J Med 318 (24): 1567-72, 1988.
- Ozaki T, Schaefer KL, Wai D, et al.: Genetic imbalances revealed by comparative genomic hybridization in osteosarcomas. Int J Cancer 102 (4): 355-65, 2002.
- Feugeas O, Guriec N, Babin-Boilletot A, et al.: Loss of heterozygosity of the RB gene is a poor prognostic factor in patients with osteosarcoma. J Clin Oncol 14 (2): 467-72, 1996.
- Heinsohn S, Evermann U, Zur Stadt U, et al.: Determination of the prognostic value of loss of heterozygosity at the retinoblastoma gene in osteosarcoma. Int J Oncol 30 (5): 1205-14, 2007.
- Goto A, Kanda H, Ishikawa Y, et al.: Association of loss of heterozygosity at the p53 locus with chemoresistance in osteosarcomas. Jpn J Cancer Res 89 (5): 539-47, 1998.
- Serra M, Pasello M, Manara MC, et al.: May P-glycoprotein status be used to stratify high-grade osteosarcoma patients? Results from the Italian/Scandinavian Sarcoma Group 1 treatment protocol. Int J Oncol 29 (6): 1459-68, 2006.
- Pakos EE, Ioannidis JP: The association of P-glycoprotein with response to chemotherapy and clinical outcome in patients with osteosarcoma. A meta-analysis. Cancer 98 (3): 581-9, 2003.
- Schwartz CL, Gorlick R, Teot L, et al.: Multiple drug resistance in osteogenic sarcoma: INT0133 from the Children's Oncology Group. J Clin Oncol 25 (15): 2057-62, 2007.
Características genómicas del osteosarcoma
Características moleculares del osteosarcoma
El panorama genómico del osteosarcoma es distinto al de otros tipos de cáncer infantil. Se caracteriza por un número inusual de variantes estructurales y un número relativamente pequeño de variantes de un solo nucleótido, en comparación con muchos cánceres en adultos.[1,2]
Las observaciones clave relacionadas con el panorama genómico del osteosarcoma son las siguientes:
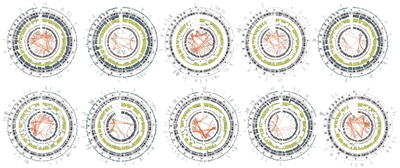
- El número de variantes estructurales observadas en el osteosarcoma es alto: más de 200 variantes estructurales por genoma;[1,2] por lo tanto, el osteosarcoma exhibe el genoma más caótico de los cánceres infantiles. Los diagramas Circos que se presentan en la Figura ilustran los números excepcionalmente altos de translocaciones intra e intercromosómicas que caracterizan los genomas del osteosarcoma.
- La carga mutacional del tumor (TMB) en niños y adolescentes con osteosarcoma es de alrededor de 2 mutaciones por megabase y es más alta que la de otros tipos de cáncer infantil (por ejemplo, sarcoma de Ewing o tumores rabdoides).[1,2] Sin embargo, esa tasa es muy inferior a la de los cánceres en adultos, como el melanoma o el cáncer de pulmón de células no pequeñas, que responden a los inhibidores de puntos de control.
- En lugar de variantes activadoras en los oncogenes y variantes inactivadoras de los genes supresores de tumores, como se observa en muchos tipos de cáncer, el panorama genómico del osteosarcoma se debe a la ganancia o amplificación del número de copias en regiones cromosómicas que incluyen pérdida (deleciones) de oncogenes y número de copias en regiones cromosómicas que incluyen genes supresores de tumores. A continuación, se describen las ganancias y pérdidas recurrentes del número de copias que afectan los oncogenes y los genes supresores de tumores conocidos, respectivamente.
Las estimaciones de la frecuencia con la que se presentan alteraciones genómicas específicas en el osteosarcoma varían de un informe a otro. Este hallazgo quizás se deba a las diferentes definiciones que se usan para describir las alteraciones en el número de copias, los distintos métodos que se utilizan para su detección o las diferencias en las características biológicas del tumor entre las poblaciones de pacientes (por ejemplo, recién diagnosticado versus en recaída, localizado versus metastásico o pediátrico versus adulto).
- En la mayoría de los casos de osteosarcoma hay alteraciones genómicas en el gen TP53 que conducen a la pérdida de función de TP53.[1] Una forma característica de inactivación de TP53 se produce por variantes estructurales en el primer intrón de TP53 que causan la alteración del gen TP53.[1] También se observan otros mecanismos de inactivación de TP53, como las mutaciones de aminoácido y las mutaciones interruptoras, así como las deleciones del gen TP53.[1,2] La combinación de varios de estos mecanismos que originan la pérdida de función de TP53 produce una inactivación bialélica en la mayoría de los casos de osteosarcoma. Debido a que muchas de las variaciones estructurales que conducen a la inactivación de TP53 se detectan mejor a través de la secuenciación del genoma completo, los resultados con base en paneles de pruebas genómicas clínicas tal vez muestren tasas más bajas de alteraciones de TP53 porque no detectan dichos cambios.[3]
- La amplificación de MDM2, que es otra alteración genómica que conduce a la pérdida de función de TP53, se observa en una minoría de casos de osteosarcoma (alrededor de un 5 %).[1,2,3,4]
- El gen RB1 se inactiva con frecuencia en el osteosarcoma, a veces por variantes deletéreas, pero de manera más habitual por deleción cromosómica de la región del cromosoma 13q14 que incluye RB1.[1,2,5]
- Las deleciones cromosómicas que incluyen al cromosoma 9p21 llevan a la deleción del gen CDN2A en alrededor del 20 % de los casos de osteosarcoma.[1,2,5]
- Entre los oncogenes tumorales, el gen MYC en el cromosoma 8q24 muestra ganancia o amplificación en cerca del 10 % de los pacientes.[3,5,6] En un estudio se relacionó la ganancia o amplificación de MYC con un pronóstico inferior. En un segundo estudio, la ganancia o amplificación de MYC fue más frecuente en niños que en adultos.[6]
- El gen CCNE1 en el cromosoma 19q12 es otro oncogén tumoral que muestra ganancia o amplificación en alrededor del 10 % de los pacientes.[3,5,6] Otras regiones cromosómicas con oncogén que muestran ganancia o amplificación cromosómica en una minoría de casos de osteosarcoma son la región que alberga el gen CDK4 en el cromosoma 12q14,[4,5,7] las regiones que albergan los genes VEGFA y CCND3 en el cromosoma 6p12,[3,4,5,7] la región que alberga el gen CCND1 en el cromosoma 11q13[4] y las regiones que albergan los genes PDGFRA, KIT y KDR en el cromosoma 4q12.[3,4,5]
- El alargamiento alternativo de los telómeros (ALT) es el mecanismo de mantenimiento de telómeros empleado por la mayoría de los tumores de osteosarcoma.[1,8,9] Las variantes inactivadoras del gen ATRX y las deleciones génicas se relacionan con el mecanismo de mantenimiento de telómeros ALT. Las alteraciones genómicas de ATRX están presentes en un subconjunto de tumores de osteosarcoma que usan este mecanismo de mantenimiento de telómeros.[1,3,9]
- Muchas de las alteraciones genómicas notificadas para los tumores de osteosarcoma en el momento del diagnóstico no proporcionan dianas terapéuticas evidentes, ya que reflejan la pérdida de genes supresores de tumores (por ejemplo, los genes TP53, RB1 o PTEN), en lugar de la activación de oncogenes que pueden servir como dianas terapéuticas. Además, el éxito ha sido limitado en los diagnósticos de cáncer al usar las ganancias o amplificaciones de los oncogenes relevantes en el osteosarcoma para identificar a los pacientes que quizás se beneficien de la terapia dirigida.
Predisposición genética al osteosarcoma
Las variantes germinales de varios genes se relacionan con la susceptibilidad al osteosarcoma. En el Cuadro 2 se resumen los síndromes y los genes asociados con estas afecciones. En un reciente estudio genómico multiinstitucional de más de 1200 pacientes con osteosarcoma, se detectaron variantes germinales patógenas, o probablemente patógenas, en genes de susceptibilidad al cáncer de herencia autosómica dominante en un 18 % de los pacientes. Estos genes de susceptibilidad al cáncer fueron más frecuentes en niños de 10 años o menores.[10]
Variantes deTP53
Las variantes del gen TP53 son las alteraciones más comunes de la línea germinal relacionadas con el osteosarcoma. Se encuentran mutaciones en este gen en cerca del 70 % de los pacientes con síndrome de Li-Fraumeni (SLF), que se relaciona con un mayor riesgo de osteosarcoma, cáncer de mama, varios tipos de cáncer de encéfalo (incluso el cerebro), sarcomas de tejido blando y otros cánceres. Aunque el rabdomiosarcoma es el sarcoma más común en pacientes de 5 años o menos que tienen SLF relacionado con TP53, el osteosarcoma es el sarcoma más común en niños y adolescentes de 6 a 19 años.[11] En un estudio se observó una frecuencia alta de casos de osteosarcoma en jóvenes (edad <30 años) que albergaban una variante conocida en el gen TP53 relacionada con el SLF o posiblemente relacionada con dicho síndrome (3,8 %), o que albergaban una variante exónica rara en TP53 (5,7 %); la frecuencia general de variante de TP53 fue del 9,5 %.[12] En otros grupos se notificaron tasas más bajas (3–7 %) de variantes de la línea germinal del gen TP53 en los pacientes con osteosarcoma.[10,13,14]
Variantes deRECQL4
Los investigadores analizaron datos de secuenciación del exoma completo de la línea germinal de 4435 pacientes con cáncer pediátrico del St. Jude Children's Research Hospital y 1127 pacientes de la base de datos National Cancer Institute's Therapeutically Applicable Research to Generate Effective Treatment (TARGET). Se identificaron 24 pacientes (0,43 %) que albergaban variantes de pérdida de función de RECQL4, entre ellos, 5 de 249 pacientes (2,0 %) con osteosarcoma.[15] Estas variantes en RECQL4 estaban significativamente sobrerrepresentadas en niños con osteosarcoma, el cáncer que se observa más a menudo en los pacientes con síndrome de Rothmund-Thomson, en comparación con los 134 187 controles sin cáncer de la base de datos Genome Aggregation Database (gnomAD v2.1; P = 0,00087; oportunidad relativa, 7,1; intervalo de confianza 95 %, 2,9–17). De las 24 personas mencionadas antes, 9 (38 %) exhibían la misma variante c.1573delT (p.Cys525Alafs) en el dominio altamente conservado de la helicasa de DNA, lo que indica que una alteración en este dominio es central para la oncogénesis.
| Síndrome | Descripción | Localización | Gen | Función |
|---|---|---|---|---|
| LMA = leucemia mieloide aguda; IL-1 = interleucina-1; SMD = síndrome mielodisplásico; RANKL = ligando del receptor activador del factor nuclear κ β; FNT = factor de necrosis tumoral. | ||||
| a Adaptación de Kansara et al.[16] | ||||
| Síndrome de Bloom[17] | Es un trastorno hereditario raro caracterizado por estatura baja y cambios por fotosensibilidad cutánea. A menudo se manifiesta con cara alargada y angosta, maxilar inferior pequeño, nariz grande y orejas prominentes. | 15q26.1 | BLM | Helicasa de DNA |
| Anemia de Diamond-Blackfan[18] | Aplasia pura de células rojas hereditaria. Pacientes en riesgo de SMD y LMA. Se relaciona con anomalías esqueléticas, como rasgos faciales anormales (dorso nasal plano e hipertelorismo ocular). | Proteínas ribosómicas | Producción ribosómica[18,19] | |
| Síndrome de Li-Fraumeni[20] | Variante hereditaria del genTP53. Los familiares afectados tienen un aumento del riesgo de tumores óseos, cáncer de mama, leucemia, tumores de encéfalo y sarcomas. | 17p13.1 | TP53 | Reacción al daño del DNA |
| Enfermedad de Paget[21] | Osteolisis excesiva con formación y remodelación de hueso anormal, lo que produce dolor por debilidad y deformidad óseas. | 18q21-qa22 | LOH18CR1 | Señalización IL-1/FNT; vía de señalización RANKL |
| 5q31 | ||||
| 5q35-qter | ||||
| Retinoblastoma[22] | Tumor maligno de retina Cerca del 66 % de los pacientes recibe el diagnóstico a los 2 años de edad y el 95 % lo recibe a los 3 años. Los pacientes con variantes hereditarias en las células germinales presentan un riesgo más alto de neoplasias subsiguientes. | 13q14.2 | RB1 | Punto de control del ciclo celular |
| Síndrome de Rothmund-Thomson (también llamado poiquilodermia congénita)[23,24] | Afección autosómica recesiva. Presenta manifestaciones cutáneas (atrofia, telangiectasias y pigmentación), vello escaso, cataratas, estatura baja y anormalidades esqueléticas. Aumento en la incidencia de osteosarcoma a menor edad. | 8q24.3 | RECQL4 | Helicasa de DNA |
| Síndrome de Werner[25] | Los pacientes a menudo tienen estatura baja, y un poco después de los 20 años aparecen signos de envejecimiento, como encanecimiento y esclerosis cutánea. Más adelante presentan otros problemas propios del envejecimiento como cataratas, úlceras cutáneas y aterosclerosis. | 8p12-p11.2 | WRN | Helicasa de DNA; actividad de exonucleasa |
Para obtener más información sobre estos síndromes genéticos, consulte los siguientes resúmenes:
- Genética del cáncer de piel (síndrome de Bloom, síndrome de Rothmund-Thomson y síndrome de Werner).
El siguiente resumen solo está disponible en inglés:
Referencias:
- Chen X, Bahrami A, Pappo A, et al.: Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7 (1): 104-12, 2014.
- Perry JA, Kiezun A, Tonzi P, et al.: Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 111 (51): E5564-73, 2014.
- Marinoff AE, Spurr LF, Fong C, et al.: Clinical Targeted Next-Generation Panel Sequencing Reveals MYC Amplification Is a Poor Prognostic Factor in Osteosarcoma. JCO Precis Oncol 7: e2200334, 2023.
- Suehara Y, Alex D, Bowman A, et al.: Clinical Genomic Sequencing of Pediatric and Adult Osteosarcoma Reveals Distinct Molecular Subsets with Potentially Targetable Alterations. Clin Cancer Res 25 (21): 6346-6356, 2019.
- Nacev BA, Sanchez-Vega F, Smith SA, et al.: Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat Commun 13 (1): 3405, 2022.
- De Noon S, Ijaz J, Coorens TH, et al.: MYC amplifications are common events in childhood osteosarcoma. J Pathol Clin Res 7 (5): 425-431, 2021.
- Parsons DW, Janeway KA, Patton DR, et al.: Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients With Refractory Cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol 40 (20): 2224-2234, 2022.
- Sanders RP, Drissi R, Billups CA, et al.: Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 22 (18): 3790-7, 2004.
- de Nonneville A, Salas S, Bertucci F, et al.: TOP3A amplification and ATRX inactivation are mutually exclusive events in pediatric osteosarcomas using ALT. EMBO Mol Med 14 (10): e15859, 2022.
- Mirabello L, Zhu B, Koster R, et al.: Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol 6 (5): 724-734, 2020.
- Ognjanovic S, Olivier M, Bergemann TL, et al.: Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 118 (5): 1387-96, 2012.
- Mirabello L, Yeager M, Mai PL, et al.: Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 107 (7): , 2015.
- Toguchida J, Yamaguchi T, Dayton SH, et al.: Prevalence and spectrum of germline mutations of the p53 gene among patients with sarcoma. N Engl J Med 326 (20): 1301-8, 1992.
- McIntyre JF, Smith-Sorensen B, Friend SH, et al.: Germline mutations of the p53 tumor suppressor gene in children with osteosarcoma. J Clin Oncol 12 (5): 925-30, 1994.
- Maciaszek JL, Oak N, Chen W, et al.: Enrichment of heterozygous germline RECQL4 loss-of-function variants in pediatric osteosarcoma. Cold Spring Harb Mol Case Stud 5 (5): , 2019.
- Kansara M, Thomas DM: Molecular pathogenesis of osteosarcoma. DNA Cell Biol 26 (1): 1-18, 2007.
- German J: Bloom's syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet 93 (1): 100-6, 1997.
- Lipton JM, Federman N, Khabbaze Y, et al.: Osteogenic sarcoma associated with Diamond-Blackfan anemia: a report from the Diamond-Blackfan Anemia Registry. J Pediatr Hematol Oncol 23 (1): 39-44, 2001.
- Idol RA, Robledo S, Du HY, et al.: Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis 39 (1): 35-43, 2007 Jul-Aug.
- Li FP, Fraumeni JF, Mulvihill JJ, et al.: A cancer family syndrome in twenty-four kindreds. Cancer Res 48 (18): 5358-62, 1988.
- Grimer RJ, Cannon SR, Taminiau AM, et al.: Osteosarcoma over the age of forty. Eur J Cancer 39 (2): 157-63, 2003.
- Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
- Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
- Hicks MJ, Roth JR, Kozinetz CA, et al.: Clinicopathologic features of osteosarcoma in patients with Rothmund-Thomson syndrome. J Clin Oncol 25 (4): 370-5, 2007.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
Clasificación celular del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo
El osteosarcoma es un tumor maligno que se caracteriza por la formación directa de hueso o tejido osteoide por parte de las células tumorales. En la clasificación histológica de la Organización Mundial de la Salud [1] de los tumores óseos, los osteosarcomas se dividen en tumores centrales (medulares) y tumores superficiales (periféricos) [2,3] y se reconocen varios subtipos dentro de cada grupo.
Tumores centrales (medulares)
- Osteosarcomas centrales convencionales. El subtipo patológico más común es el osteosarcoma central convencional, que se caracteriza por áreas de necrosis, mitosis atípicas, y tejido osteoide o cartilaginoso maligno. Los otros subtipos son mucho menos comunes y cada uno se presenta con una frecuencia de menos del 5 %.
- Osteosarcomas telangiectásicos.[4,5] El osteosarcoma telangiectásico a veces se confunde en la radiografía con un quiste aneurismático de hueso o un tumor de células gigantes. Esta variante se debe abordar como un osteosarcoma convencional.[4,5]
- Osteosarcomas intraóseos bien diferenciados (grado bajo).
- Osteosarcomas de células pequeñas.
Tumores superficiales (periféricos)
Los términos osteosarcoma parosteal y periosteal están incorporados en la bibliografía y son de uso amplio. Sin embargo, son confusos para los pacientes y los médicos. Sería más útil dividir el osteosarcoma por su localización y grado histológico. El osteosarcoma de grado alto, que a veces se llama osteosarcoma convencional, por lo habitual surge en el centro y crece hacia el exterior, destruye la corteza y los tejidos blandos circundantes; no obstante, hay casos inequívocos de osteosarcoma de grado alto superficiales.[6] De manera similar, hay informes de osteosarcoma de grado bajo que surge en la cavidad medular.
- Osteosarcomas parosteales (yuxtacorticales) bien diferenciados (grado bajo).[7,8] El osteosarcoma parosteal se define como una lesión que surge de la superficie del hueso con un aspecto bien diferenciado en las imágenes y características histológicas de grado bajo.[9] El sitio más común del osteosarcoma parosteal es la parte posterior y distal del fémur. El osteosarcoma parosteal es más frecuente en pacientes mayores que el osteosarcoma convencional de grado alto, que es más común en pacientes de 20 a 30 años. Se puede tratar con éxito mediante la escisión amplia del tumor primario solo.[7,10]
- Osteosarcomas periosteales (osteosarcomas de grado bajo a intermedio).[11,12,13] El osteosarcoma periosteal suele aparecer como una masa de tejido blando de base amplia con erosión extrínseca de la corteza ósea subyacente.[12] Las características patológicas indican un grado intermedio de diferenciación. La resección amplia es esencial.
En una revisión retrospectiva de una sola institución, se identificó a 29 pacientes con osteosarcoma periosteal.[11] La tasa de supervivencia sin enfermedad a 5 años fue del 83 %. Los autores no pudieron emitir una conclusión definitiva sobre los beneficios de la quimioterapia adyuvante.
En otra revisión retrospectiva de una sola institución, se identificó a 33 pacientes con osteosarcoma periosteal.[13] La tasa de supervivencia general (SG) a 10 años fue del 84 %. La tasa de SG a 10 años fue del 83 % para los pacientes tratados con cirugía sola y del 86 % para los pacientes tratados con cirugía y quimioterapia.
La European Musculoskeletal Oncology Society hizo un análisis retrospectivo de 119 pacientes con osteosarcoma periosteal; 17 pacientes tenían metástasis.[12] La tasa de SG fue del 89 % a los 5 años y del 83 % a los 10 años. De los pacientes, 81 recibieron quimioterapia y 50 de ellos la recibieron antes de la resección quirúrgica definitiva. No hubo diferencia en los desenlaces de los pacientes que recibieron quimioterapia y los que no la recibieron.
- Osteosarcomas superficiales de grado alto.[3,6,14]
Osteosarcoma extraóseo
El osteosarcoma extraóseo es una neoplasia mesenquimatosa maligna sin vínculo directo con el sistema esquelético. En épocas anteriores, el tratamiento del osteosarcoma extraóseo seguía las mismas directrices que el tratamiento del sarcoma de tejido blando;[15] no obstante, en un análisis retrospectivo del Cooperative German-Austrian-Swiss Osteosarcoma Study Group, se identificó un desenlace favorable para los pacientes con osteosarcoma extraóseo tratados con cirugía y el tratamiento del osteosarcoma convencional.[16]
Sarcoma pleomórfico indiferenciado óseo
El sarcoma pleomórfico indiferenciado (SPI) óseo se debe diferenciar del histiocitoma fibroso angiomatoide, un tumor de grado bajo que no suele ser invasor, es pequeño y se relaciona con un desenlace excelente cuando se trata con cirugía sola.[17] En un estudio se indican las tasas de supervivencia sin complicaciones similares para el SPI y el osteosarcoma.[18]
Referencias:
- Schajowicz F, Sissons HA, Sobin LH: The World Health Organization's histologic classification of bone tumors. A commentary on the second edition. Cancer 75 (5): 1208-14, 1995.
- Antonescu CR, Huvos AG: Low-grade osteogenic sarcoma arising in medullary and surface osseous locations. Am J Clin Pathol 114 (Suppl): S90-103, 2000.
- Kaste SC, Fuller CE, Saharia A, et al.: Pediatric surface osteosarcoma: clinical, pathologic, and radiologic features. Pediatr Blood Cancer 47 (2): 152-62, 2006.
- Bacci G, Ferrari S, Ruggieri P, et al.: Telangiectatic osteosarcoma of the extremity: neoadjuvant chemotherapy in 24 cases. Acta Orthop Scand 72 (2): 167-72, 2001.
- Weiss A, Khoury JD, Hoffer FA, et al.: Telangiectatic osteosarcoma: the St. Jude Children's Research Hospital's experience. Cancer 109 (8): 1627-37, 2007.
- Okada K, Unni KK, Swee RG, et al.: High grade surface osteosarcoma: a clinicopathologic study of 46 cases. Cancer 85 (5): 1044-54, 1999.
- Hoshi M, Matsumoto S, Manabe J, et al.: Oncologic outcome of parosteal osteosarcoma. Int J Clin Oncol 11 (2): 120-6, 2006.
- Han I, Oh JH, Na YG, et al.: Clinical outcome of parosteal osteosarcoma. J Surg Oncol 97 (2): 146-9, 2008.
- Kumar VS, Barwar N, Khan SA: Surface osteosarcomas: Diagnosis, treatment and outcome. Indian J Orthop 48 (3): 255-61, 2014.
- Schwab JH, Antonescu CR, Athanasian EA, et al.: A comparison of intramedullary and juxtacortical low-grade osteogenic sarcoma. Clin Orthop Relat Res 466 (6): 1318-22, 2008.
- Rose PS, Dickey ID, Wenger DE, et al.: Periosteal osteosarcoma: long-term outcome and risk of late recurrence. Clin Orthop Relat Res 453: 314-7, 2006.
- Grimer RJ, Bielack S, Flege S, et al.: Periosteal osteosarcoma--a European review of outcome. Eur J Cancer 41 (18): 2806-11, 2005.
- Cesari M, Alberghini M, Vanel D, et al.: Periosteal osteosarcoma: a single-institution experience. Cancer 117 (8): 1731-5, 2011.
- Staals EL, Bacchini P, Bertoni F: High-grade surface osteosarcoma: a review of 25 cases from the Rizzoli Institute. Cancer 112 (7): 1592-9, 2008.
- Wodowski K, Hill DA, Pappo AS, et al.: A chemosensitive pediatric extraosseous osteosarcoma: case report and review of the literature. J Pediatr Hematol Oncol 25 (1): 73-7, 2003.
- Goldstein-Jackson SY, Gosheger G, Delling G, et al.: Extraskeletal osteosarcoma has a favourable prognosis when treated like conventional osteosarcoma. J Cancer Res Clin Oncol 131 (8): 520-6, 2005.
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
- Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
Información sobre la estadificación y el sitio del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo
Tradicionalmente, se utilizaba el sistema de estadificación de Enneking para las neoplasias malignas esqueléticas con el fin de estadificar el osteosarcoma y el SPI óseo.[1] Con este sistema se infería el grado de malignidad del tumor primario usando los descriptores intracompartimental o extracompartimental. El sistema de estadificación TNM (tumor, ganglio linfático y metástasis) del American Joint Committee on Cancer para los tumores óseos malignos no se emplea de forma generalizada para el osteosarcoma infantil, y los pacientes no se estratifican a partir de grupos de estadio pronóstico.
Para el tratamiento, el osteosarcoma se define según las siguientes categorías:
- Localizado. Se considera que los pacientes sin enfermedad metastásica clínicamente detectable tienen un osteosarcoma localizado.
- Metastásico. Se considera que los pacientes con metástasis en cualquier sitio detectable mediante estudios clínicos de rutina en el momento de la presentación inicial tienen osteosarcoma metastásico.
Osteosarcoma localizado
Los tumores localizados se circunscriben al hueso de origen. Se considera que los pacientes con lesiones discontinuas confinadas al mismo hueso del tumor primario tienen una enfermedad localizada siempre y cuando las lesiones discontinuas se puedan incluir en una resección quirúrgica planificada.[2] Alrededor de la mitad de los tumores se originan en el fémur y, de estos, el 80 % se presentan en la porción distal del fémur. Otros sitios primarios, en orden descendiente de frecuencia, son la porción proximal de la tibia, la porción proximal del húmero, la pelvis, el maxilar, el peroné y las costillas.[3] Es más probable que el osteosarcoma de cabeza y cuello sea de grado bajo [4] y que se presente en pacientes de más edad que el osteosarcoma en el esqueleto apendicular.
Osteosarcoma metastásico
Se encuentran indicios radiológicos de depósitos tumorales metastásicos en alrededor del 20 % de los pacientes en el momento del diagnóstico; del 85 % al 90 % de la enfermedad metastásica es pulmonar. El segundo sitio más común de metástasis es otro hueso; este tipo de metástasis puede ser solitaria o múltiple.[5]
El síndrome de osteosarcoma multifocal se refiere a la aparición de múltiples focos de osteosarcoma sin un tumor primario evidente, a menudo con compromiso metafisario simétrico.[3]
Evaluación para la estadificación
En los pacientes con osteosarcoma confirmado se obtienen radiografías simples del sitio primario que abarcan una proyección de un solo plano de todo el hueso afectado para evaluar metástasis discontinuas; también se deben llevar a cabo estudios de estadificación antes del tratamiento como los siguientes:[6]
- Imágenes por resonancia magnética (IRM) del sitio primario para incluir todo el hueso.
- Tomografía computarizada (TC) si no se dispone de IRM.
- Tomografía por emisión de positrones (TEP) con flúor F 18-fludesoxiglucosa (18F-FDG)-TC o TEP-IRM.[7,8]
- Gammagrafía ósea si la TEP no está disponible.
- Radiografía del tórax posteroanterior y lateral.
- TC del tórax.
En una revisión retrospectiva de 206 pacientes con osteosarcoma, se comparó la utilidad de la gammagrafía ósea, la TEP y la TEP-TC para la detección de metástasis óseas.[9] La TEP-TC fue más sensible y precisa que la gammagrafía ósea (sensibilidad del 92 vs. el 74 %), y el uso combinado de ambos estudios de imágenes logró la sensibilidad más alta para el diagnóstico de metástasis óseas en el osteosarcoma (100 %). La TEP con 18F-FDG es la modalidad de estadificación preferida para la detección de lesiones óseas. La TC sigue siendo necesaria para evaluar la metástasis pulmonar.
Referencias:
- Enneking WF: A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res (204): 9-24, 1986.
- Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
- Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
- Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
- Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
- Meyer JS, Nadel HR, Marina N, et al.: Imaging guidelines for children with Ewing sarcoma and osteosarcoma: a report from the Children's Oncology Group Bone Tumor Committee. Pediatr Blood Cancer 51 (2): 163-70, 2008.
- Oh C, Bishop MW, Cho SY, et al.: 18F-FDG PET/CT in the Management of Osteosarcoma. J Nucl Med 64 (6): 842-851, 2023.
- Quartuccio N, Fox J, Kuk D, et al.: Pediatric bone sarcoma: diagnostic performance of ¹⁸F-FDG PET/CT versus conventional imaging for initial staging and follow-up. AJR Am J Roentgenol 204 (1): 153-60, 2015.
- Byun BH, Kong CB, Lim I, et al.: Comparison of (18)F-FDG PET/CT and (99 m)Tc-MDP bone scintigraphy for detection of bone metastasis in osteosarcoma. Skeletal Radiol 42 (12): 1673-81, 2013.
Aspectos generales de las opciones de tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo
Es imprescindible que un oncólogo ortopédico familiarizado con el abordaje quirúrgico de esta enfermedad haga la evaluación inicial de los pacientes en quienes se sospecha o se confirmó la presencia de un osteosarcoma. Esta evaluación, que incluye estudios con imágenes, se debe hacer antes de la biopsia inicial, porque una biopsia que se lleve a cabo de manera incorrecta suele perjudicar la posibilidad de hacer un procedimiento con conservación del miembro. Además, se recomienda el uso de soportes protectores para los pacientes con tumores en huesos que soportan carga a fin de evitar fracturas patológicas que impidan la cirugía para conservar el miembro.
Por lo general, un tratamiento exitoso exige la combinación de quimioterapia sistémica eficaz y una resección completa de la enfermedad clínicamente detectable.
En ensayos clínicos aleatorizados se ha establecido que tanto la quimioterapia adyuvante como la quimioterapia neoadyuvante son eficaces para prevenir las recaídas en pacientes con tumores no metastásicos en la evaluación clínica.[1]; [2][Nivel de evidencia A1] El Pediatric Oncology Group (POG) condujo un estudio en el que los pacientes se asignaron al azar para una amputación inmediata o una amputación después de la terapia neoadyuvante. Un gran porcentaje de pacientes rehusó someterse a la asignación aleatoria y el estudio se suspendió sin lograr alcanzar las metas fijadas para el número de participantes. En el pequeño grupo de pacientes que llegaron a tratarse, no hubo ninguna diferencia en los desenlaces entre quienes recibieron quimioterapia preoperatoria y los que recibieron quimioterapia posoperatoria.[3]
El tratamiento del osteosarcoma también depende del grado histológico, como se explica a continuación:
- Osteosarcoma de grado bajo. Los pacientes con osteosarcoma de grado bajo a veces se tratan con éxito mediante una resección quirúrgica amplia, con independencia del sitio de origen.
- Osteosarcoma de grado intermedio. Los patólogos describen algunos tumores como osteosarcomas de grado intermedio. Es difícil tomar decisiones sobre el tratamiento de los pacientes con osteosarcomas de grado intermedio. Cuando una biopsia tumoral indica que hay un osteosarcoma de grado intermedio, una opción es proceder con una resección amplia. Si se obtiene el tumor completo, esto permite que el patólogo examine más tejido, evalúe el tejido blando y la invasión linfovascular lo que, a menudo, aclara la naturaleza de la lesión.
Si se confirma que la lesión contiene elementos de grado alto, se indica quimioterapia sistémica, al igual que para cualquier osteosarcoma de grado alto. El POG dirigió un estudio en pacientes con osteosarcoma de grado alto que se asignaron al azar a cirugía definitiva inmediata seguida de quimioterapia adyuvante o un período inicial de quimioterapia seguida de cirugía definitiva.[3] El desenlace fue el mismo para ambos grupos. Aunque la estrategia de quimioterapia inicial seguida de cirugía definitiva se ha convertido en el abordaje de aplicación casi universal para el osteosarcoma, en este estudio se indica que no aumenta el riesgo de fracaso del tratamiento si se realiza la cirugía definitiva antes de la quimioterapia; esto quizás sirva para aclarar los diagnósticos equívocos del osteosarcoma de grado intermedio.
- Osteosarcoma de grado alto. Los pacientes con osteosarcoma de grado alto requieren cirugía y quimioterapia sistémica, ya sea que el tumor se origine en un lugar central convencional o en la superficie ósea.
Es importante identificar el osteosarcoma intraóseo bien diferenciado y el osteosarcoma parosteal, porque los pacientes con estos tipos de tumores tienen un pronóstico más favorable y es posible tratarlos de manera eficaz solo con una extirpación amplia del tumor primario.[4,5] En general, los pacientes con osteosarcoma periosteal tienen un pronóstico favorable [6] y el tratamiento se orienta a partir del grado histológico.[5,7]
Los pacientes con sarcoma pleomórfico indiferenciado (SPI) óseo se tratan de acuerdo con los protocolos de tratamiento del osteosarcoma.[8] Se notificó una tasa de supervivencia específica del sarcoma del 70,7 % con regímenes a base, principalmente, de cisplatino y doxorrubicina.[9]
Las modalidades de imágenes, como la resonancia magnética dinámica o la tomografía por emisión de positrones, son métodos no invasivos para evaluar la respuesta,[10,11,12,13,14,15,16,17,18] y son las modalidades preferidas en el ensayo AOST2032 (NCT05691478) del Children's Oncology Group.
En el Cuadro 3 se enumeran las opciones de tratamiento para el osteosarcoma y el SPI óseo localizados, metastásicos y recidivantes.
Referencias:
- Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
- Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
- Goorin AM, Schwartzentruber DJ, Devidas M, et al.: Presurgical chemotherapy compared with immediate surgery and adjuvant chemotherapy for nonmetastatic osteosarcoma: Pediatric Oncology Group Study POG-8651. J Clin Oncol 21 (8): 1574-80, 2003.
- Hoshi M, Matsumoto S, Manabe J, et al.: Oncologic outcome of parosteal osteosarcoma. Int J Clin Oncol 11 (2): 120-6, 2006.
- Schwab JH, Antonescu CR, Athanasian EA, et al.: A comparison of intramedullary and juxtacortical low-grade osteogenic sarcoma. Clin Orthop Relat Res 466 (6): 1318-22, 2008.
- Rose PS, Dickey ID, Wenger DE, et al.: Periosteal osteosarcoma: long-term outcome and risk of late recurrence. Clin Orthop Relat Res 453: 314-7, 2006.
- Grimer RJ, Bielack S, Flege S, et al.: Periosteal osteosarcoma--a European review of outcome. Eur J Cancer 41 (18): 2806-11, 2005.
- Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
- Veitch ZW, Fasih S, Griffin AM, et al.: Clinical outcomes of non-osteogenic, non-Ewing soft-tissue sarcoma of bone--experience of the Toronto Sarcoma Program. Cancer Med 9 (24): 9282-9292, 2020.
- Reddick WE, Wang S, Xiong X, et al.: Dynamic magnetic resonance imaging of regional contrast access as an additional prognostic factor in pediatric osteosarcoma. Cancer 91 (12): 2230-7, 2001.
- Hawkins DS, Conrad EU, Butrynski JE, et al.: [F-18]-fluorodeoxy-D-glucose-positron emission tomography response is associated with outcome for extremity osteosarcoma in children and young adults. Cancer 115 (15): 3519-25, 2009.
- Cheon GJ, Kim MS, Lee JA, et al.: Prediction model of chemotherapy response in osteosarcoma by 18F-FDG PET and MRI. J Nucl Med 50 (9): 1435-40, 2009.
- Costelloe CM, Macapinlac HA, Madewell JE, et al.: 18F-FDG PET/CT as an indicator of progression-free and overall survival in osteosarcoma. J Nucl Med 50 (3): 340-7, 2009.
- Hamada K, Tomita Y, Inoue A, et al.: Evaluation of chemotherapy response in osteosarcoma with FDG-PET. Ann Nucl Med 23 (1): 89-95, 2009.
- Bajpai J, Kumar R, Sreenivas V, et al.: Prediction of chemotherapy response by PET-CT in osteosarcoma: correlation with histologic necrosis. J Pediatr Hematol Oncol 33 (7): e271-8, 2011.
- Kong CB, Byun BH, Lim I, et al.: ¹⁸F-FDG PET SUVmax as an indicator of histopathologic response after neoadjuvant chemotherapy in extremity osteosarcoma. Eur J Nucl Med Mol Imaging 40 (5): 728-36, 2013.
- Byun BH, Kong CB, Lim I, et al.: Combination of 18F-FDG PET/CT and diffusion-weighted MR imaging as a predictor of histologic response to neoadjuvant chemotherapy: preliminary results in osteosarcoma. J Nucl Med 54 (7): 1053-9, 2013.
- Davis JC, Daw NC, Navid F, et al.: 18F-FDG Uptake During Early Adjuvant Chemotherapy Predicts Histologic Response in Pediatric and Young Adult Patients with Osteosarcoma. J Nucl Med 59 (1): 25-30, 2018.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es raro, aunque desde 1975 se ha observado un aumento gradual de la incidencia general.[1] Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes profesionales de atención de la salud y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatra.
- Cirujanos de trasplante.
- Patólogos.
- Radioncólogos pediatras.
- Oncólogos y hematólogos pediatras.
- Oftalmólogos.
- Especialistas en rehabilitación.
- Enfermeros especializados en oncología pediátrica.
- Trabajadores o asistentes sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas y dietistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes con cáncer infantil.[2] En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
Los niños y adolescentes sobrevivientes de cáncer necesitan una vigilancia estrecha, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan, o se presenten, meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Referencias:
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo localizados
Los pacientes con osteosarcoma localizado que se someten a cirugía y quimioterapia presentan una tasa de supervivencia general (SG) a 5 años del 62 % al 65 %.[1] Es muy importante llevar a cabo una resección quirúrgica completa en los pacientes con osteosarcoma localizado; aunque esta no es suficiente como terapia única. Por lo menos el 80 % de los pacientes tratados con cirugía sola presentarán enfermedad metastásica.[2] En ensayos clínicos aleatorizados se estableció que la quimioterapia adyuvante es eficaz para prevenir la recaída o la recidiva en pacientes con tumores primarios resecables localizados.[2]; [3][Nivel de evidencia A1]
El sarcoma pleomórfico indiferenciado óseo (SPI) es más común en adultos mayores. Los pacientes con SPI óseo se tratan de acuerdo con los protocolos terapéuticos para el osteosarcoma. El desenlace de los pacientes con SPI resecable es similar al desenlace de los pacientes con osteosarcoma.[4] Del mismo modo que para el osteosarcoma, los pacientes con un grado de necrosis favorable (≥90 % de necrosis) tienen una supervivencia más prolongada que aquellos con un grado de necrosis inferior (<90 % necrosis).[5] Muchos pacientes con SPI necesitarán quimioterapia preoperatoria para lograr una escisión local amplia.[6]
Opciones de tratamiento para el osteosarcoma y el sarcoma pleomórfico indiferenciado óseo localizados
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo localizados son las siguientes:
- Extirpación quirúrgica del tumor primario.
- Quimioterapia (antes o después de la resección quirúrgica definitiva del tumor primario).
- Radioterapia, si no es posible realizar una cirugía o si los márgenes quirúrgicos son inadecuados.
Extirpación quirúrgica del tumor primario
La extirpación quirúrgica del tumor primario con márgenes adecuados es un componente esencial de la estrategia curativa en los pacientes con osteosarcoma localizado. El tipo de cirugía necesaria para la ablación completa del tumor primario depende de una serie de factores que se deben evaluar caso por caso.[7]
Procedimientos con conservación del miembro
En general, más del 80 % de los pacientes con osteosarcoma de extremidades se pueden tratar mediante cirugía para conservar un miembro y no necesitan amputación.[8] Los procedimientos con conservación del miembro se planifican solo cuando la estadificación preoperatoria indica que es posible lograr márgenes quirúrgicos amplios. En un estudio, los pacientes sometidos a procedimientos para conservar un miembro que tuvieron una respuesta histológica deficiente y márgenes quirúrgicos estrechos presentaron una tasa alta de recidiva local.[9]
Hay muchas opciones para la reconstrucción después de la cirugía para conservar un miembro, entre ellas, el uso de endoprótesis metálicas, aloinjertos, injerto óseo autógeno vascularizado y rotoplastia. Otra opción es la distracción osteogénica con transporte óseo para los pacientes cuyos tumores no comprometen la epífisis de huesos largos.[10] Con este procedimiento se logra una reconstrucción estable que restablece el funcionamiento normal de la extremidad.
La elección de la reconstrucción quirúrgica óptima implica muchos factores; entre ellos, los siguientes:[11][Nivel de evidencia A1]
- Sitio y tamaño del tumor primario.
- Capacidad para preservar el suministro neurovascular a la porción distal de la extremidad.
- Edad del paciente y posibilidad de crecimiento adicional.
- Necesidades y deseos del paciente y la familia para desempeñar funciones específicas como la participación en deportes.
Si una reconstrucción complicada retrasa o prohíbe la reanudación de la quimioterapia sistémica, es posible que la conservación del miembro ponga en peligro la probabilidad de curación. En análisis retrospectivos de 703 pacientes con osteosarcoma localizado, la reanudación de la quimioterapia 21 días o más después de la cirugía definitiva se relacionó con un aumento del riesgo de muerte y efectos adversos (cociente de riesgos instantáneos [CRI], 1,57; 1,04–2,36).[11] Las demoras en el periodo hasta que se completa la quimioterapia también se relacionaron con una supervivencia general (SG) y una supervivencia sin complicaciones (SSC) más bajas. En un análisis multivariante retrospectivo de 113 pacientes con osteosarcoma localizado, una demora en completar la quimioterapia superior a 4 semanas se relacionó con un CRI de la SG de 2,70 (1,11–6,76, P = 0,003) y un CRI de SSC de 1,13 (1,00–1,26, P = 0,016).[12]
Amputación
En algunos pacientes, la amputación sigue siendo la mejor opción para el tratamiento del tumor primario. Una fractura patológica en el momento del diagnóstico o durante la quimioterapia preoperatoria no excluye la cirugía para conservar un miembro cuando se pueden obtener márgenes quirúrgicos amplios.[13] Si el examen patológico de la muestra quirúrgica muestra márgenes inadecuados, se debe considerar una amputación inmediata, en especial si la necrosis histológica después de la quimioterapia preoperatoria fue precaria.[14]
Factores relacionados con un aumento del riesgo de recidiva local
Los pacientes sometidos a amputación tienen tasas de recidiva local más bajas que los pacientes sometidos a procedimientos para conservar un miembro.[15] Sin embargo, no hay diferencia en la SG entre los pacientes tratados al inicio con amputación y quienes se sometieron a un procedimiento con conservación del miembro. Los pacientes con tumores en el fémur tienen una tasa de recidiva local más alta que aquellos con tumores primarios en la tibia o el peroné. Se ha evaluado la rotoplastia y otros procedimientos de conservación de un miembro en cuanto a su resultado funcional y efecto en la supervivencia. Si bien la resección con conservación del miembro es el procedimiento vigente para el control local en la mayoría de las instituciones pediátricas, hay pocos datos que indiquen que la conservación del miembro inferior es superior a la amputación con respecto a la calidad de vida de los pacientes.[16]
El Cooperative Osteosarcoma Study Group (COSS) alemán condujo un análisis retrospectivo de 1802 pacientes con osteosarcoma localizado y metastásico sometidos a resección quirúrgica de toda la enfermedad clínicamente detectable.[17][Nivel de evidencia C1] La recidiva local (n = 76) se relacionó con un riesgo alto de muerte por osteosarcoma. Los factores relacionados con un aumento del riesgo de recidiva local incluyeron la ausencia de participación en un ensayo clínico, sitio primario en la pelvis, cirugía para conservar un miembro, infiltración del tejido blando más allá del periostio, respuesta patológica precaria a la quimioterapia inicial, incumplimiento de la quimioterapia planificada y obtención de la biopsia en una institución diferente a la institución donde se realizará la cirugía definitiva.
Quimioterapia
Quimioterapia preoperatoria
Casi todos los pacientes reciben quimioterapia preoperatoria intravenosa como tratamiento inicial. Sin embargo, no se ha determinado ningún régimen de quimioterapia estándar. Los protocolos actuales de quimioterapia incluyen combinaciones de los siguientes fármacos: dosis altas de metotrexato, doxorrubicina, ciclofosfamida, cisplatino, ifosfamida, etopósido y carboplatino.[18,19,20,21,22,23,24,25,26]
Evidencia (quimioterapia preoperatoria):
- En un metanálisis de protocolos para el tratamiento del osteosarcoma se obtuvieron las siguientes conclusiones:[27]
- Los regímenes de 3 fármacos quimioterapéuticos fueron superiores a los regímenes con 2 fármacos activos.
- Los regímenes de 4 fármacos activos no fueron superiores a los regímenes de 3 fármacos activos.
- Los regímenes de 3 fármacos que no incluyeron dosis altas de metotrexato fueron inferiores a los regímenes de 3 fármacos que incluyeron dosis altas de metotrexato.
- En un estudio italiano en el que se usaron regímenes con menos cursos de dosis altas de metotrexato se observaron los siguientes resultados:[28][Nivel de evidencia B4]
- La probabilidad de SSC fue inferior que en estudios anteriores en los que se usaron regímenes con más cursos de dosis altas de metotrexato.
- El Children's Oncology Group (COG) llevó a cabo un ensayo aleatorizado prospectivo de niños y adultos jóvenes con osteosarcoma localizado recién diagnosticado. Todos los pacientes recibieron cisplatino, doxorrubicina y dosis altas de metotrexato. La mitad de los pacientes se asignaron de manera aleatoria para recibir ifosfamida. En una segunda aleatorización, la mitad de los pacientes se asignaron a recibir el compuesto biológico muramil tripéptido fosfatidiletanolamina encapsulado en liposomas (L-MTP-PE) después de la resección quirúrgica definitiva. Se observó lo siguiente:[29][Nivel de evidencia A1]
- La adición de ifosfamida no mejoró el desenlace.
- La adición de L-MTP-PE produjo una mejora de la tasa de SSC, que no superó la prueba convencional de significación estadística (P = 0,08), y una mejora significativa de la tasa de SG (78 vs. 70 %; P = 0,03).
- Se ha especulado sobre la posible contribución del tratamiento posrecaída, aunque no se identificaron diferencias en los abordajes quirúrgicos posrecaída en los pacientes que recayeron. La utilidad de L-MTP-PE en el tratamiento del osteosarcoma sigue siendo tema de discusión.[30]
- El COG realizó una serie de estudios piloto en pacientes con osteosarcoma localizado recién diagnosticado.[31][Nivel de evidencia B4]
- En el estudio piloto 1, los pacientes con grados más bajos de necrosis después del tratamiento inicial con 3 fármacos recibieron terapia subsiguiente con una dosis acumulada de doxorrubicina de 600 mg/m2.
- En el estudio piloto 2, todos los pacientes recibieron quimioterapia inicial de 4 fármacos con cisplatino, doxorrubicina, dosis altas de metotrexato e ifosfamida. Los pacientes con los grados más bajos de necrosis recibieron quimioterapia subsiguiente con una dosis acumulada de doxorrubicina superior a 600 mg/m2.
- En el estudio piloto 3, todos los pacientes recibieron la misma quimioterapia inicial de 4 fármacos que los pacientes del estudio piloto 2. Los pacientes con grados más bajos de necrosis recibieron dosis más altas de ifosfamida con la adición de etopósido durante el tratamiento posterior.
Los resultados de estos estudios se enumeran a continuación:
- Los desenlaces de los tres estudios piloto fueron similares entre sí y a los de los controles históricos.
- Todos los pacientes recibieron dexrazoxano antes de cada dosis de doxorrubicina. La adición de dexrazoxano no pareció disminuir la tasa de necrosis favorable después del tratamiento inicial ni redujo la SSC.
- La fracción de acortamiento ventricular izquierda, medida con ecocardiografía, se modificó muy poco a las 78 semanas del ingreso al estudio.
- No se obtuvo evidencia de aumento del riesgo de una leucemia secundaria.
- El consorcio internacional del European and American Osteosarcoma Study Group (EURAMOS) se conformó con el fin de conducir un gran ensayo prospectivo aleatorizado para determinar si la modificación del régimen de quimioterapia según el grado de necrosis mejoraría la SSC. Todos los pacientes recibieron tratamiento inicial con cisplatino, doxorrubicina y dosis altas de metotrexato (MAP). Los pacientes con más del 90 % de necrosis se asignaron al azar para continuar con la misma quimioterapia luego de la cirugía o recibir la misma quimioterapia con adición de interferón α-2b. Los pacientes con menos del 90 % de necrosis se asignaron al azar para continuar con la misma quimioterapia o recibir la misma quimioterapia con adición de dosis altas de ifosfamida y etopósido (MAPIE).[32][Nivel de evidencia A1]
- Al cabo de una mediana de seguimiento de 54 meses para todos los pacientes registrados (N = 2260), la tasa de SSC a 3 años fue del 59 % (intervalo de confianza [IC] 95 %, 57–61 %), y la tasa de SSC a 5 años fue del 54 % (IC 95 %, 52–56 %).
- La tasa de SG a 3 años fue del 79 % (IC 95 %, 77–81 %), y la tasa de SG a 5 años fue del 71 % (IC 95 %, 68–73 %).
- Los pacientes con enfermedad localizada en el momento del diagnóstico (M0) sometidos a resección quirúrgica completa (n = 1549) presentaron tasas de SSC y SG a 3 años desde la cirugía del 70 % y el 88 %, respectivamente; las tasas de SSC y SG a 5 años desde la cirugía fueron del 64 % y el 79 %, respectivamente.
- De los pacientes o familias participantes en el estudio, el 40 % declinó la aleatorización después de la resección quirúrgica definitiva, lo que dificultó la interpretación de las respuestas a las preguntas del estudio aleatorizado y cuestionó las posibilidad de generalización de los resultados.
- De los pacientes con más del 90 % de necrosis, 716 se asignaron al azar para continuar con la misma quimioterapia luego de la cirugía, con adición del interferón α-2b, o sin este. Las tasas de SSC a 3 años fueron del 74 % (IC 95 %, 69–79 %) para los pacientes que recibieron MAP y del 77 % (IC 95 %, 72–81 %) para los pacientes que recibieron MAP con interferón α-2b. El CRI fue de 0,83 (IC 95 %, 0,61–1,12; P = 0,2).[33]
- De los pacientes con menos del 90 % de necrosis, 618 se asignaron al azar a continuar con quimioterapia MAP o recibir quimioterapia MAPIE. Las estimaciones de la SSC a 3 años para los pacientes con enfermedad localizada fueron del 60 % (IC 95 %, 54–66 %) para el grupo de MAP, y del 57 % (IC 95 %, 51–63 %) para el grupo de MAPIE. El CRI fue de 1,03 (0,81–1,33, P = 0,80).[34]
- En un ensayo del Sarcoma Group francés, se suprimieron las dosis altas de metotrexato en los pacientes adultos con osteosarcoma. En la cohorte se incluyeron 60 pacientes de 16 a 63 años (mediana de edad, 27 años). Los pacientes recibieron tratamiento inicial con ifosfamida y cisplatino. Se añadió doxorrubicina después de la cirugía definitiva en los pacientes que presentaron menos necrosis después de la quimioterapia inicial.[35]
- Al cabo de una mediana de seguimiento de 322 meses, la tasa de supervivencia sin enfermedad (SSE) a 5 años fue del 51,8 % y la tasa de SG a 5 años fue del 64,4 %.
- A los 10 años, la tasa de SSE fue del 49,9 % y la tasa de SG fue del 64,4 %.
- Al cabo de 25 años, la tasa de SSE fue del 47,8 % y la tasa de SG fue del 55,9 %.
Quimioterapia posoperatoria
Tradicionalmente, en algunos ensayos clínicos se usó el grado de necrosis tumoral para determinar el tipo de quimioterapia posoperatoria. En general, si la necrosis tumoral excedía el 90 %, se continuaba con el régimen de quimioterapia preoperatoria. Cuando la necrosis tumoral era menor del 90 %, algunos grupos incorporaban fármacos no utilizados en la terapia preoperatoria.
Los pacientes con menos necrosis después de la quimioterapia inicial tienen un pronóstico inferior al de los pacientes con más necrosis. Aun así, el pronóstico es mucho mejor que el de los pacientes tratados con cirugía sola sin quimioterapia adyuvante.
A partir de la evidencia que sigue, es inadecuado concluir que los pacientes con menos necrosis no responden a la quimioterapia y que la quimioterapia adyuvante se debe omitir en estos pacientes. La quimioterapia después de la cirugía definitiva debe incluir los fármacos utilizados en la primera fase del tratamiento a menos que se presente una enfermedad progresiva clara e inequívoca durante la fase inicial del tratamiento.
Evidencia (uso de los mismos fármacos durante la quimioterapia posoperatoria):
- En los primeros informes del Memorial Sloan-Kettering Cancer Center (MSKCC) se indicó que la adición de cisplatino a la quimioterapia posoperatoria mejoraba el desenlace en los pacientes con menos del 90 % de necrosis tumoral.[36]
- Con un seguimiento más largo, el desenlace para los pacientes con menos del 90 % de necrosis tumoral tratados en el MSKCC fue el mismo, ya fuera que recibieran o no cisplatino en la fase posoperatoria de tratamiento.[37]
- En una experiencia anterior, el COSS alemán de osteosarcoma dirigió un ensayo en el que se cambió el régimen de quimioterapia después del tratamiento inicial para los pacientes con poca necrosis.[38] Se interrumpió la administración de los fármacos usados antes de la cirugía y se los reemplazó con otros.
- Los desenlaces fueron mucho más precarios para estos pacientes que para los pacientes que continuaron recibiendo los mismos fármacos.
- En ensayos posteriores dirigidos por otros grupos no se logró demostrar una mejora de la SSC cuando los fármacos no incluidos en el régimen preoperatorio (cisplatino) se agregaron a la terapia posoperatoria.[19,39]
- En un ensayo piloto de una institución se probó la estrategia de interrumpir la administración de los fármacos usados durante la fase inicial para los pacientes con el grado de necrosis más precario; la terapia posoperatoria incluyó melfalán con reconstitución de células madre autógenas.[40]
- La tasa de SSC a 5 años para este grupo fue del 28 %, que fue inferior a las tasas de SSC observadas en muchas series grandes en las que se continuaron administrando los fármacos pese a un grado de necrosis más baja.
- El consorcio internacional del grupo EURAMOS se conformó con el fin de conducir un gran ensayo prospectivo aleatorizado para determinar si la modificación del régimen de quimioterapia según el grado de necrosis mejoraría la SSC. Todos los pacientes recibieron tratamiento inicial con cisplatino, doxorrubicina y dosis altas de metotrexato (MAP). Los pacientes con más del 90 % de necrosis se asignaron al azar para continuar con la misma quimioterapia luego de la cirugía o recibir la misma quimioterapia con adición de interferón α-2b.[33][Nivel de evidencia B1] En el mismo ensayo del grupo EURAMOS, los pacientes con menos del 90 % de necrosis se asignaron al azar para continuar con la misma quimioterapia o recibir la misma quimioterapia con la adición de dosis altas de ifosfamida y etopósido (MAPIE).[34][Nivel de evidencia B1]
- La adición del interferón α-2b no mejoró la probabilidad de SSC.
- Al cabo de una mediana de seguimiento de más de 61 meses, no hubo diferencias en la SSC entre los dos grupos.
- La intensificación del tratamiento en el grupo que recibió MAPIE produjo más efectos tóxicos que el tratamiento en el grupo estándar que recibió MAP.
Progresión antes del tratamiento local
En un análisis retrospectivo de una sola institución se notificó la progresión temprana del osteosarcoma antes del control local.[41] Entre 195 pacientes de 18 años o menos, 25 (81 %) presentaron progresión solo en sitios locales y 6 presentaron progresión combinada en sitios locales y metastásicos. Los autores no identificaron de manera prospectiva a pacientes con características clínicas que pudieran indicar osteosarcoma telangiectásico con aumento de necrosis y hemorragia, lo que tal vez explicaría la progresión aparente. Para toda la cohorte, la tasa de SSC a 5 años fue del 27,2 % y la tasa de SG fue del 31,3 %. Los pacientes con necrosis favorable tuvieron mejores tasas de SSC y SG a 5 años (66,7 % en ambos casos), en comparación con los pacientes con una respuesta histológica deficiente (21,4 y 25,6 %, respectivamente). No obstante, estos resultados no alcanzaron significación estadística (P = 0,07 y P = 0,1).
Otros abordajes quimioterapéuticos que no se consideran eficaces
El Italian Sarcoma Group y el Scandinavian Sarcoma Group realizaron un ensayo clínico con pacientes de osteosarcoma que presentaban enfermedad metastásica clínicamente detectable.[42] La consolidación con dosis altas de etopósido y carboplatino seguida de reconstitución de células madre autógenas no mejoró el desenlace y los investigadores no recomiendan esta estrategia para el tratamiento del osteosarcoma.
En estudios de laboratorio en los que se emplearon líneas celulares y xenoinjertos, se indicó que los bisfosfonatos tienen un efecto activo contra el osteosarcoma.[43] En un ensayo clínico de una sola institución se demostró que el pamidronato se podría administrar de manera simultánea con quimioterapia multifarmacológica a pacientes con osteosarcoma recién diagnosticado.[43] Los grupos cooperativos franceses de sarcoma en niños y adultos realizaron un ensayo prospectivo para el tratamiento del osteosarcoma.[44] Todos los pacientes recibieron quimioterapia multifarmacológica y se asignaron de manera aleatoria para recibir zoledronato o no recibirlo. La adición de zoledronato no mejoró la SSC.
Radioterapia
Si la resección quirúrgica completa no es factible, o si los márgenes quirúrgicos son inadecuados, es posible que la radioterapia mejore la tasa de control local.[45,46]; [47][Nivel de evidencia C1] La radioterapia se debe considerar para los pacientes con osteosarcoma de cabeza y cuello que tienen márgenes de resección con compromiso tumoral positivo o incierto.[48][Nivel de evidencia C1]
Evidencia (radioterapia para el control local):
- Si bien se acepta la resección quirúrgica primaria como el abordaje estándar, en un análisis retrospectivo de un grupo pequeño de pacientes cuidadosamente seleccionados, se notificó que algunos pacientes lograron una SSC a largo plazo con radioterapia externa para el control local.[49][Nivel de evidencia C1]
- Investigadores de una sola institución informaron sobre 28 niños y adultos jóvenes con osteosarcoma que recibieron radioterapia para el control local. De este grupo, 16 pacientes recibieron radioterapia durante el primer curso de tratamiento y 12 pacientes recibieron radioterapia después de la recidiva.[50]
- En los pacientes que recibieron la radioterapia durante el tratamiento primario, la incidencia acumulada de fracaso local a 5 años fue del 25 %.
- En los pacientes con enfermedad recidivante, la incidencia acumulada de fracaso local a 5 años fue del 44 %.
- Se observó progresión tumoral local en 3 de 13 pacientes (23 %) tratados con radioterapia adyuvante después de la resección, mientras que 3 de 6 pacientes (50 %) que recibieron radioterapia definitiva como modalidad única de control local, presentaron progresión local.
Osteosarcoma de cabeza y cuello
El osteosarcoma que surge en la cabeza y el cuello se presenta en una población de mayor edad que la del osteosarcoma de extremidades.[48,51,52,53,54] En el grupo de edad pediátrica, es más probable que los osteosarcomas de cabeza y cuello sean de grado bajo o intermedio, en comparación con los tumores de las extremidades.[55,56] En todas las series notificadas se destaca la necesidad de una resección quirúrgica completa.[48,51,52,53,54,55,56][Nivel de evidencia C1] La probabilidad de cura del osteosarcoma de cabeza y cuello con cirugía sola es mayor que la del osteosarcoma de extremidades. Cuando los márgenes quirúrgicos son positivos, hay una tendencia hacia la mejora de la supervivencia con radioterapia adyuvante.[48,53][Nivel de evidencia C1]
Si bien no se cuenta con ensayos aleatorizados en los que se evalúe la eficacia de la quimioterapia en pacientes con osteosarcoma de cabeza y cuello, en varias series se indica que hay un beneficio.[51,57] Se debe considerar la quimioterapia para los pacientes más jóvenes con osteosarcomas de cabeza y cuello de grado alto.[55,58]
Los pacientes con osteosarcoma de cabeza y cuello tienen un riesgo más alto de recidiva local y un riesgo más bajo de metástasis a distancia que los pacientes con osteosarcoma de extremidades.[51,53,54,59]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- Link MP, Goorin AM, Miser AW, et al.: The effect of adjuvant chemotherapy on relapse-free survival in patients with osteosarcoma of the extremity. N Engl J Med 314 (25): 1600-6, 1986.
- Bernthal NM, Federman N, Eilber FR, et al.: Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. Cancer 118 (23): 5888-93, 2012.
- Picci P, Bacci G, Ferrari S, et al.: Neoadjuvant chemotherapy in malignant fibrous histiocytoma of bone and in osteosarcoma located in the extremities: analogies and differences between the two tumors. Ann Oncol 8 (11): 1107-15, 1997.
- Bramwell VH, Steward WP, Nooij M, et al.: Neoadjuvant chemotherapy with doxorubicin and cisplatin in malignant fibrous histiocytoma of bone: A European Osteosarcoma Intergroup study. J Clin Oncol 17 (10): 3260-9, 1999.
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
- Grimer RJ: Surgical options for children with osteosarcoma. Lancet Oncol 6 (2): 85-92, 2005.
- Bacci G, Ferrari S, Bertoni F, et al.: Long-term outcome for patients with nonmetastatic osteosarcoma of the extremity treated at the istituto ortopedico rizzoli according to the istituto ortopedico rizzoli/osteosarcoma-2 protocol: an updated report. J Clin Oncol 18 (24): 4016-27, 2000.
- Grimer RJ, Taminiau AM, Cannon SR, et al.: Surgical outcomes in osteosarcoma. J Bone Joint Surg Br 84 (3): 395-400, 2002.
- Watanabe K, Tsuchiya H, Yamamoto N, et al.: Over 10-year follow-up of functional outcome in patients with bone tumors reconstructed using distraction osteogenesis. J Orthop Sci 18 (1): 101-9, 2013.
- Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
- Vasquez L, Silva J, Chavez S, et al.: Prognostic impact of diagnostic and treatment delays in children with osteosarcoma. Pediatr Blood Cancer 67 (4): e28180, 2020.
- Scully SP, Ghert MA, Zurakowski D, et al.: Pathologic fracture in osteosarcoma : prognostic importance and treatment implications. J Bone Joint Surg Am 84-A (1): 49-57, 2002.
- Bacci G, Ferrari S, Lari S, et al.: Osteosarcoma of the limb. Amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br 84 (1): 88-92, 2002.
- Reddy KI, Wafa H, Gaston CL, et al.: Does amputation offer any survival benefit over limb salvage in osteosarcoma patients with poor chemonecrosis and close margins? Bone Joint J 97-B (1): 115-20, 2015.
- Ottaviani G, Robert RS, Huh WW, et al.: Functional, psychosocial and professional outcomes in long-term survivors of lower-extremity osteosarcomas: amputation versus limb salvage. Cancer Treat Res 152: 421-36, 2009.
- Andreou D, Bielack SS, Carrle D, et al.: The influence of tumor- and treatment-related factors on the development of local recurrence in osteosarcoma after adequate surgery. An analysis of 1355 patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. Ann Oncol 22 (5): 1228-35, 2011.
- Fuchs N, Bielack SS, Epler D, et al.: Long-term results of the co-operative German-Austrian-Swiss osteosarcoma study group's protocol COSS-86 of intensive multidrug chemotherapy and surgery for osteosarcoma of the limbs. Ann Oncol 9 (8): 893-9, 1998.
- Provisor AJ, Ettinger LJ, Nachman JB, et al.: Treatment of nonmetastatic osteosarcoma of the extremity with preoperative and postoperative chemotherapy: a report from the Children's Cancer Group. J Clin Oncol 15 (1): 76-84, 1997.
- Bacci G, Picci P, Avella M, et al.: Effect of intra-arterial versus intravenous cisplatin in addition to systemic adriamycin and high-dose methotrexate on histologic tumor response of osteosarcoma of the extremities. J Chemother 4 (3): 189-95, 1992.
- Cassano WF, Graham-Pole J, Dickson N: Etoposide, cyclophosphamide, cisplatin, and doxorubicin as neoadjuvant chemotherapy for osteosarcoma. Cancer 68 (9): 1899-902, 1991.
- Voûte PA, Souhami RL, Nooij M, et al.: A phase II study of cisplatin, ifosfamide and doxorubicin in operable primary, axial skeletal and metastatic osteosarcoma. European Osteosarcoma Intergroup (EOI). Ann Oncol 10 (10): 1211-8, 1999.
- Ferrari S, Smeland S, Mercuri M, et al.: Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. J Clin Oncol 23 (34): 8845-52, 2005.
- Zalupski MM, Rankin C, Ryan JR, et al.: Adjuvant therapy of osteosarcoma--A Phase II trial: Southwest Oncology Group study 9139. Cancer 100 (4): 818-25, 2004.
- Meyers PA, Schwartz CL, Krailo M, et al.: Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol 23 (9): 2004-11, 2005.
- Daw NC, Neel MD, Rao BN, et al.: Frontline treatment of localized osteosarcoma without methotrexate: results of the St. Jude Children's Research Hospital OS99 trial. Cancer 117 (12): 2770-8, 2011.
- Anninga JK, Gelderblom H, Fiocco M, et al.: Chemotherapeutic adjuvant treatment for osteosarcoma: where do we stand? Eur J Cancer 47 (16): 2431-45, 2011.
- Ferrari S, Meazza C, Palmerini E, et al.: Nonmetastatic osteosarcoma of the extremity. Neoadjuvant chemotherapy with methotrexate, cisplatin, doxorubicin and ifosfamide. An Italian Sarcoma Group study (ISG/OS-Oss). Tumori 100 (6): 612-9, 2014 Nov-Dec.
- Meyers PA, Schwartz CL, Krailo MD, et al.: Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children's Oncology Group. J Clin Oncol 26 (4): 633-8, 2008.
- Anderson PM, Meyers P, Kleinerman E, et al.: Mifamurtide in metastatic and recurrent osteosarcoma: a patient access study with pharmacokinetic, pharmacodynamic, and safety assessments. Pediatr Blood Cancer 61 (2): 238-44, 2014.
- Schwartz CL, Wexler LH, Krailo MD, et al.: Intensified Chemotherapy With Dexrazoxane Cardioprotection in Newly Diagnosed Nonmetastatic Osteosarcoma: A Report From the Children's Oncology Group. Pediatr Blood Cancer 63 (1): 54-61, 2016.
- Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
- Bielack SS, Smeland S, Whelan JS, et al.: Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. J Clin Oncol 33 (20): 2279-87, 2015.
- Marina NM, Smeland S, Bielack SS, et al.: Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol 17 (10): 1396-1408, 2016.
- Blay JY, Penel N, Toulmonde M, et al.: Long term survival in adult osteosarcoma patients treated with a two-drug regimen: Final results of the OSAD93 phase II study of the FSG-GETO. Eur J Cancer 208: 114228, 2024.
- Rosen G, Caparros B, Huvos AG, et al.: Preoperative chemotherapy for osteogenic sarcoma: selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer 49 (6): 1221-30, 1982.
- Meyers PA, Heller G, Healey J, et al.: Chemotherapy for nonmetastatic osteogenic sarcoma: the Memorial Sloan-Kettering experience. J Clin Oncol 10 (1): 5-15, 1992.
- Winkler K, Beron G, Delling G, et al.: Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J Clin Oncol 6 (2): 329-37, 1988.
- Smeland S, Müller C, Alvegard TA, et al.: Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J Cancer 39 (4): 488-94, 2003.
- Venkatramani R, Murray J, Helman L, et al.: Risk-Based Therapy for Localized Osteosarcoma. Pediatr Blood Cancer 63 (3): 412-7, 2016.
- Halalsheh H, Amer S, Sultan I: Progression before local control in osteosarcoma: Outcome and prognosis-predictive factors. Pediatr Blood Cancer 70 (11): e30649, 2023.
- Boye K, Del Prever AB, Eriksson M, et al.: High-dose chemotherapy with stem cell rescue in the primary treatment of metastatic and pelvic osteosarcoma: final results of the ISG/SSG II study. Pediatr Blood Cancer 61 (5): 840-5, 2014.
- Meyers PA, Healey JH, Chou AJ, et al.: Addition of pamidronate to chemotherapy for the treatment of osteosarcoma. Cancer 117 (8): 1736-44, 2011.
- Piperno-Neumann S, Le Deley MC, Rédini F, et al.: Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol 17 (8): 1070-80, 2016.
- Ozaki T, Flege S, Kevric M, et al.: Osteosarcoma of the pelvis: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 21 (2): 334-41, 2003.
- DeLaney TF, Park L, Goldberg SI, et al.: Radiotherapy for local control of osteosarcoma. Int J Radiat Oncol Biol Phys 61 (2): 492-8, 2005.
- Ciernik IF, Niemierko A, Harmon DC, et al.: Proton-based radiotherapy for unresectable or incompletely resected osteosarcoma. Cancer 117 (19): 4522-30, 2011.
- Guadagnolo BA, Zagars GK, Raymond AK, et al.: Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. Cancer 115 (14): 3262-70, 2009.
- Hundsdoerfer P, Albrecht M, Rühl U, et al.: Long-term outcome after polychemotherapy and intensive local radiation therapy of high-grade osteosarcoma. Eur J Cancer 45 (14): 2447-51, 2009.
- Tinkle CL, Lu J, Han Y, et al.: Curative-intent radiotherapy for pediatric osteosarcoma: The St. Jude experience. Pediatr Blood Cancer 66 (8): e27763, 2019.
- Canadian Society of Otolaryngology-Head and Neck Surgery Oncology Study Group: Osteogenic sarcoma of the mandible and maxilla: a Canadian review (1980-2000). J Otolaryngol 33 (3): 139-44, 2004.
- Kassir RR, Rassekh CH, Kinsella JB, et al.: Osteosarcoma of the head and neck: meta-analysis of nonrandomized studies. Laryngoscope 107 (1): 56-61, 1997.
- Laskar S, Basu A, Muckaden MA, et al.: Osteosarcoma of the head and neck region: lessons learned from a single-institution experience of 50 patients. Head Neck 30 (8): 1020-6, 2008.
- Patel SG, Meyers P, Huvos AG, et al.: Improved outcomes in patients with osteogenic sarcoma of the head and neck. Cancer 95 (7): 1495-503, 2002.
- Gadwal SR, Gannon FH, Fanburg-Smith JC, et al.: Primary osteosarcoma of the head and neck in pediatric patients: a clinicopathologic study of 22 cases with a review of the literature. Cancer 91 (3): 598-605, 2001.
- Daw NC, Mahmoud HH, Meyer WH, et al.: Bone sarcomas of the head and neck in children: the St Jude Children's Research Hospital experience. Cancer 88 (9): 2172-80, 2000.
- Smeele LE, Snow GB, van der Waal I: Osteosarcoma of the head and neck: meta-analysis of the nonrandomized studies. Laryngoscope 108 (6): 946, 1998.
- Smeele LE, Kostense PJ, van der Waal I, et al.: Effect of chemotherapy on survival of craniofacial osteosarcoma: a systematic review of 201 patients. J Clin Oncol 15 (1): 363-7, 1997.
- Jasnau S, Meyer U, Potratz J, et al.: Craniofacial osteosarcoma Experience of the cooperative German-Austrian-Swiss osteosarcoma study group. Oral Oncol 44 (3): 286-94, 2008.
Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo metastásicos en el momento del diagnóstico
Cerca del 20 % al 25 % de los pacientes con osteosarcoma exhiben una enfermedad metastásica clínicamente detectable. Entre los pacientes con enfermedad metastásica en el momento de la presentación inicial, alrededor del 20 % permanecerá sin enfermedad y alrededor del 30 % sobrevivirá 5 años a partir del momento del diagnóstico.[1]
Los pulmones son los sitios más comunes de enfermedad metastásica inicial.[2] Los pacientes con metástasis limitadas a los pulmones tienen un mejor desenlace que los pacientes con metástasis en otros sitios o en los pulmones combinadas con metástasis en otros sitios.[1,3]
Opciones de tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo metastásicos en el momento del diagnóstico
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo metastásicos en el momento del diagnóstico son las siguientes:
- Quimioterapia.
Los fármacos quimioterapéuticos usados incluyen el metotrexato en dosis altas, la doxorrubicina, el cisplatino, la ifosfamida en dosis altas, el etopósido y, en algunos informes, el carboplatino o la ciclofosfamida.
Evidencia (quimioterapia):
- En un ensayo se investigó el uso de dosis altas de ifosfamida (17,5 g por curso) en combinación con etopósido para los pacientes con osteosarcoma metastásico recién diagnosticado; se observaron los siguientes resultados:[4]
- La combinación produjo una respuesta completa en el 10 % de los pacientes y una respuesta parcial en el 49 % de los pacientes.
Sin embargo, del mismo modo que para la enfermedad localizada, no hay evidencia de que la adición de ifosfamida o etopósido contribuya a mejorar la supervivencia sin complicaciones (SSC) o la supervivencia general (SG) de los pacientes con enfermedad metastásica.
- En un estudio de diseño factorial de pacientes con osteosarcoma metastásico (n = 91) se evaluó la adición de muramil tripéptido o ifosfamida al régimen de quimioterapia estándar que incluyó cisplatino, dosis altas de metotrexato y doxorrubicina.[5]
- Hubo una ventaja nominal con la adición de muramil tripéptido (aunque no con la de ifosfamida) en términos de SSC y de SG, pero no se cumplieron los criterios de significación estadística.
- En un grupo del consorcio internacional European and American Osteosarcoma Study (EURAMOS), 362 de 2186 pacientes (17 %) presentaban metástasis en el momento del diagnóstico. Los pacientes se asignaron de manera aleatoria para recibir tratamiento con cisplatino, doxorrubicina y dosis altas de metotrexato, o cisplatino, doxorrubicina, dosis altas de metotrexato e ifosfamida.[6][Nivel de evidencia A1]
- Al cabo de una mediana de seguimiento de 47 meses, la tasa de SSC a 3 años fue del 32 % (intervalo de confianza [IC] 95 %, 27–37 %), y la tasa de SSC a 5 años fue del 28 % (IC 95 %, 23–33 %).
- La tasa de SG a 3 años fue del 56 % (IC 95 %, 50–61 %), y la tasa de SG a 5 años fue del 45 % (IC 95 %, 39–50 %).
Las opciones de tratamiento del SPI óseo con metástasis en el momento de la presentación inicial son las mismas que para el osteosarcoma metastásico. Los pacientes con SPI irresecable o metastásico tienen un desenlace muy precario.[7]
Opciones de tratamiento para las metástasis pulmonares solas en el momento del diagnóstico
Las opciones de tratamiento para los pacientes con lesiones pulmonares metastásicas en el momento del diagnóstico son las siguientes:
- Quimioterapia preoperatoria seguida de cirugía para extirpar el tumor, y luego quimioterapia combinada posoperatoria.
Los pacientes con lesiones metastásicas en el pulmón como único sitio de enfermedad metastásica deben, en la medida de lo posible, someterse a la resección de las lesiones pulmonares. En general, este procedimiento se hace después de la administración de la quimioterapia preoperatoria. Luego de la resección quirúrgica definitiva del tumor primario, la mayoría de los médicos reinician la quimioterapia sistémica antes de la cirugía pulmonar para evitar demoras prolongadas en el reinicio de la quimioterapia. En cerca del 10 % de los pacientes, todas las lesiones pulmonares desaparecen después de la quimioterapia preoperatoria.[3] Es posible lograr la resección completa de la enfermedad metastásica pulmonar en un porcentaje alto de pacientes con nódulos residuales en el pulmón después de la quimioterapia preoperatoria. La supervivencia a largo plazo es precaria para los pacientes que no se someten a una resección quirúrgica completa de la enfermedad pulmonar metastásica.[8,9][Nivel de evidencia B4]
En los pacientes que presentan osteosarcoma primario y metástasis limitada a los pulmones, y que logran una remisión quirúrgica completa, la tasa de SSC a 5 años es del 20 % al 25 %. La presencia de nódulos metastásicos múltiples confiere un pronóstico más precario que la presencia de 1 o 2 nódulos, y el compromiso pulmonar bilateral es peor que el compromiso unilateral.[1] Los pacientes con lesiones pulmonares periféricas quizás tengan un mejor pronóstico que aquellos con lesiones centrales.[10] Los pacientes con menos de 3 nódulos restringidos a un pulmón tal vez logren una SSC a 5 años del 40 % al 50 % aproximadamente.[1]
En un análisis multiinstitucional retrospectivo se comparó la toracotomía con la toracoscopia para la resección de metástasis pulmonares en pacientes con osteosarcoma.[11] En el análisis se incluyeron pacientes que tenían metástasis pulmonares en el momento del diagnóstico, pacientes con recidiva pulmonar tras el tratamiento inicial de la enfermedad localizada y pacientes con progresión de la enfermedad durante el tratamiento. Los autores reconocieron un importante sesgo de selección de los pacientes elegidos para someterse a toracoscopia. En un análisis de regresión de Cox, en el que se examinaron otros factores que influyen en el desenlace, se produjo un aumento significativo del riesgo de mortalidad (cociente de riesgos instantáneos [CRI], 2,11; IC 95 %, 1,09–4,09; P = 0,027), pero no de recidiva pulmonar (CRI, 0,96; IC 95 %, 0,52–1,79; P = 0,90) con un abordaje toracoscópico. En un análisis de subconjunto limitado a los pacientes con enfermedad oligometastásica, la toracoscopia no aumentó el riesgo de mortalidad (CRI, 1,16; IC 95 %, 0,64–2,11; P = 0,62). El ensayo aleatorizado en curso (AOST2031 [NCT05235165]) se diseñó para abordar de manera definitiva esta cuestión y el sesgo de selección. En este ensayo se comparará el efecto de la toracotomía con la cirugía toracoscópica.
Opciones de tratamiento para las metástasis óseas con metástasis pulmonares o sin estas
El segundo sitio metastásico más común es un hueso alejado del tumor primario. Los pacientes con metástasis en otros huesos alejados del tumor primario tienen unas tasas de SSC y SG de casi un 10 %.[1] En un estudio de pacientes que presentaron tumores primarios en las extremidades y metástasis sincrónica en otros huesos, solo 3 de 46 pacientes permanecieron libres de enfermedad 5 años después.[12] Los pacientes con lesiones discontinuas transarticulares tienen el pronóstico más precario.[13]
El osteosarcoma multifocal es diferente al osteosarcoma que se presenta con una lesión primaria claramente definida y una metástasis ósea limitada. El osteosarcoma multifocal se presenta de manera habitual como lesiones metafisarias simétricas y, a veces, es difícil identificar la lesión primaria. Los pacientes con enfermedad ósea multifocal en el momento de la presentación inicial tienen un pronóstico muy precario, pero el tratamiento con quimioterapia sistémica y resección quirúrgica radical quizás prolongue la vida de manera significativa.[14,15]
Las opciones de tratamiento para los pacientes que presentan metástasis óseas, con metástasis pulmonares o sin estas, son las siguientes:
- Quimioterapia preoperatoria seguida de cirugía para extirpar el tumor primario. Después de la resección quirúrgica definitiva del tumor primario, la mayoría de los médicos reinician la quimioterapia sistémica.
El momento de la cirugía para extirpar los tumores metastásicos no se ha definido bien. No suele intentarse al mismo tiempo que la cirugía primaria porque los retrasos de más de 21 días en reiniciar la quimioterapia aumentan el riesgo de efectos adversos y muerte.[16]
- Cirugía para extirpar el tumor primario seguida de quimioterapia y, luego, resección de la enfermedad metastásica seguida de quimioterapia combinada posoperatoria.
Es posible utilizar un abordaje alternativo de tratamiento cuando no es posible seguir el curso habitual de tratamiento de quimioterapia preoperatoria seguida de ablación quirúrgica del tumor primario y resección de toda la enfermedad metastásica visible seguida de quimioterapia combinada posoperatoria. Este abordaje de tratamiento alternativo comienza con cirugía del tumor primario, seguida de quimioterapia y, luego, resección quirúrgica de la enfermedad metastásica. Este enfoque de tratamiento quizás sea apropiado para pacientes con dolor intratable, fractura patológica o infección incontrolada del tumor, cuando el inicio de la quimioterapia acarree riesgo de septicemia.[17]
- Resección de las lesiones óseas metastásicas si es posible.
- Radioterapia dirigida a las extremidades.
Se ha comprobado que la radioterapia dirigida a las extremidades quizás confiera algo de control local.[18]
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- AOST2031 (NCT05235165) (Thoracotomy Versus Thoracoscopic Management of Pulmonary Metastases in Patients With Osteosarcoma): en este ensayo de fase III se compara el efecto de la toracotomía con la cirugía toracoscópica (cirugía toracoscópica asistida por video) en el tratamiento de pacientes con osteosarcoma que se extendió al pulmón (metástasis pulmonar). Este ensayo se realiza para evaluar los 2 métodos quirúrgicos diferentes para estos pacientes y determinar qué procedimiento es mejor.
- AOST2032 (NCT05691478) (A Study to Test the Addition of the Drug Cabozantinib to Chemotherapy in Patients With Newly Diagnosed Osteosarcoma): este ensayo comienza con una fase de viabilidad para pacientes con osteosarcoma de riesgo alto con tumor primario resecable. Después de la fase de viabilidad se hará un estudio aleatorizado para comparar la quimioterapia que incluye metotrexato, doxorrubicina y cisplatino (MAP) con MAP y cabozantinib para pacientes con osteosarcoma metastásico localizado de diagnóstico reciente.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Kager L, Zoubek A, Pötschger U, et al.: Primary metastatic osteosarcoma: presentation and outcome of patients treated on neoadjuvant Cooperative Osteosarcoma Study Group protocols. J Clin Oncol 21 (10): 2011-8, 2003.
- Kempf-Bielack B, Bielack SS, Jürgens H, et al.: Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 23 (3): 559-68, 2005.
- Bacci G, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. J Surg Oncol 98 (6): 415-20, 2008.
- Goorin AM, Harris MB, Bernstein M, et al.: Phase II/III trial of etoposide and high-dose ifosfamide in newly diagnosed metastatic osteosarcoma: a pediatric oncology group trial. J Clin Oncol 20 (2): 426-33, 2002.
- Chou AJ, Kleinerman ES, Krailo MD, et al.: Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children's Oncology Group. Cancer 115 (22): 5339-48, 2009.
- Smeland S, Bielack SS, Whelan J, et al.: Survival and prognosis with osteosarcoma: outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur J Cancer 109: 36-50, 2019.
- Daw NC, Billups CA, Pappo AS, et al.: Malignant fibrous histiocytoma and other fibrohistiocytic tumors in pediatric patients: the St. Jude Children's Research Hospital experience. Cancer 97 (11): 2839-47, 2003.
- Yamamoto Y, Kanzaki R, Kanou T, et al.: Long-term outcomes and prognostic factors of pulmonary metastasectomy for osteosarcoma and soft tissue sarcoma. Int J Clin Oncol 24 (7): 863-870, 2019.
- Slade AD, Warneke CL, Hughes DP, et al.: Effect of concurrent metastatic disease on survival in children and adolescents undergoing lung resection for metastatic osteosarcoma. J Pediatr Surg 50 (1): 157-60; discussion 160, 2015.
- Letourneau PA, Xiao L, Harting MT, et al.: Location of pulmonary metastasis in pediatric osteosarcoma is predictive of outcome. J Pediatr Surg 46 (7): 1333-7, 2011.
- Lautz TB, Farooqui Z, Jenkins T, et al.: Thoracoscopy vs thoracotomy for the management of metastatic osteosarcoma: A Pediatric Surgical Oncology Research Collaborative Study. Int J Cancer 148 (5): 1164-1171, 2021.
- Bacci G, Fabbri N, Balladelli A, et al.: Treatment and prognosis for synchronous multifocal osteosarcoma in 42 patients. J Bone Joint Surg Br 88 (8): 1071-5, 2006.
- Kager L, Zoubek A, Kastner U, et al.: Skip metastases in osteosarcoma: experience of the Cooperative Osteosarcoma Study Group. J Clin Oncol 24 (10): 1535-41, 2006.
- Harris MB, Gieser P, Goorin AM, et al.: Treatment of metastatic osteosarcoma at diagnosis: a Pediatric Oncology Group Study. J Clin Oncol 16 (11): 3641-8, 1998.
- Longhi A, Fabbri N, Donati D, et al.: Neoadjuvant chemotherapy for patients with synchronous multifocal osteosarcoma: results in eleven cases. J Chemother 13 (3): 324-30, 2001.
- Imran H, Enders F, Krailo M, et al.: Effect of time to resumption of chemotherapy after definitive surgery on prognosis for non-metastatic osteosarcoma. J Bone Joint Surg Am 91 (3): 604-12, 2009.
- Marina NM, Smeland S, Bielack SS, et al.: Comparison of MAPIE versus MAP in patients with a poor response to preoperative chemotherapy for newly diagnosed high-grade osteosarcoma (EURAMOS-1): an open-label, international, randomised controlled trial. Lancet Oncol 17 (10): 1396-1408, 2016.
- Tinkle CL, Lu J, Han Y, et al.: Curative-intent radiotherapy for pediatric osteosarcoma: The St. Jude experience. Pediatr Blood Cancer 66 (8): e27763, 2019.
Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo recidivantes
Alrededor del 50 % de las recaídas en pacientes con osteosarcoma recidivante se presentan en el transcurso de los 18 meses tras completar el tratamiento y solo el 5 % de las recaídas se presentan después de 5 años.[1,2,3,4]
Factores pronósticos de recidiva
Los factores pronósticos del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo recidivantes son los siguientes:
- Tiempo transcurrido desde el diagnóstico. En una serie de 564 pacientes con recidiva, aquellos cuya enfermedad recidivó en el transcurso de los 2 años después del diagnóstico tuvieron un pronóstico más precario que los pacientes cuya enfermedad recidivó luego de 2 años.[1] En otra serie de 431 pacientes, las recidivas que se presentaron en un período inferior a los 2 años desde el diagnóstico también se relacionaron con desenlaces más precarios.[5]
- Edad en el momento del diagnóstico inicial. Una mayor edad en el momento de la inscripción inicial en el estudio se relacionó con un pronóstico más precario después de la recidiva.[5]
- Enfermedad metastásica en el momento del diagnóstico. La enfermedad metastásica en el momento de la presentación inicial se relacionó con un pronóstico más precario.[5]
- Respuesta tumoral a la quimioterapia preoperatoria. Los pacientes con respuesta histológica favorable a la quimioterapia preoperatoria inicial tuvieron una mejor supervivencia general (SG) luego de la recidiva que aquellos con una respuesta inicial precaria.[1]
- Sitio de las metástasis. En dos series grandes, la incidencia de recidiva según el sitio fue la siguiente: pulmón solo (65–80 %), hueso solo (8–10 %), recidiva local sola (4–7 %) y recaída combinada (10–15 %).[4,6] Las metástasis abdominales son infrecuentes, pero a veces se presentan incluso 4 años después del diagnóstico.[7] El Children's Oncology Group (COG) notificó los desenlaces de 431 pacientes jóvenes con osteosarcoma recidivante.[5][Nivel de evidencia C3] Los pacientes con recidivas pulmonares y óseas tuvieron desenlaces más precarios que los pacientes con recidivas solo en los pulmones (P = 0,005).
- Resecabilidad quirúrgica. Los pacientes con osteosarcoma recidivante se deben evaluar para determinar la posibilidad de resección quirúrgica, ya que a veces se curan con resección quirúrgica radical, con quimioterapia o sin esta.[8,6,9,10,11,12]
El control del osteosarcoma después de una recidiva depende de la resección quirúrgica completa de todos los sitios de metástasis clínica detectable. Si no se intenta la resección quirúrgica, o no es posible hacerla, entonces la progresión y la muerte son inevitables. La capacidad de lograr una resección completa de la enfermedad recidivante es el factor pronóstico más importante en el momento de la primera recaída, con una tasa de supervivencia a 5 años del 20 % al 45 % después de una resección completa de tumores pulmonares metastásicos, y una tasa de supervivencia del 20 % tras una resección completa de metástasis en otros sitios.[4,6,12,13]
Opciones de tratamiento para el osteosarcoma y el sarcoma pleomórfico indiferenciado óseo recidivantes
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo recidivantes son las siguientes:
- Cirugía para extirpar todos los sitios de enfermedad metastásica.
- Quimioterapia y terapia dirigida.
- Radiofármacos y radioterapia.
Cirugía
El control del osteosarcoma después de una recidiva depende de la resección quirúrgica completa de todos los sitios de metástasis clínica detectable.
En un análisis multiinstitucional retrospectivo se comparó la toracotomía con la toracoscopia para la resección de metástasis pulmonares en pacientes con osteosarcoma.[14] En el análisis se incluyeron pacientes que tenían metástasis pulmonares en el momento del diagnóstico, pacientes con recidiva pulmonar tras el tratamiento inicial de la enfermedad localizada y pacientes con progresión de la enfermedad durante el tratamiento. Los autores reconocieron un importante sesgo de selección de los pacientes elegidos para someterse a toracoscopia. En un análisis de regresión de Cox, en el que se examinaron otros factores que influyen en el desenlace, se produjo un aumento significativo del riesgo de mortalidad (cociente de riesgos instantáneos [CRI], 2,11; intervalo de confianza [IC] 95 %, 1,09–4,09; P = 0,027), pero no recidiva pulmonar (CRI, 0.96; IC 95 %, 0,52–1,79; P = 0,90) con un abordaje toracoscópico. En un análisis de subconjunto limitado a los pacientes con enfermedad oligometastásica, la toracoscopia no aumentó el riesgo de mortalidad (CRI, 1,16; IC 95 %, 0,64–2,11; P = 0,62). El ensayo aleatorizado en curso (AOST2031 [NCT05235165]) se diseñó para abordar de manera definitiva esta cuestión y el sesgo de selección. Este ensayo comparará el efecto de la toracotomía con la cirugía toracoscópica.
Quimioterapia y terapia dirigida
La función de la quimioterapia sistémica para el tratamiento de pacientes con osteosarcoma recidivante no está bien definida. La selección de tratamiento sistémico adicional depende de muchos factores, incluso el sitio de la recidiva, el tratamiento primario previo y las consideraciones individuales de cada paciente.
Con frecuencia, el osteosarcoma tiene una matriz estromal que se mineraliza aún más con la necrosis tumoral, dejando una masa que se observa en las imágenes y que puede tener o no un número reducido de células tumorales en su interior. Por lo tanto, es posible que los criterios estándar Response Evaluation Criteria in Solid Tumors (RECIST) no sean apropiados para evaluar la respuesta a los fármacos en pacientes con osteosarcoma. El COG, en un intento por establecer las tasas de supervivencia sin complicaciones (SSC) iniciales en pacientes con osteosarcoma en recaída, analizó los desenlaces de estos pacientes en 7 ensayos de fase II de un solo grupo. Se determinó que los fármacos analizados en cada ensayo eran inactivos según las tasas de respuesta radiográfica.[15]
- La tasa de SSC para 96 pacientes con osteosarcoma y enfermedad cuantificable fue del 12 % a los 4 meses (IC 95 %, 6–19 %).
- No hubo ninguna diferencia significativa en la SSC entre los ensayos, de acuerdo con el número de regímenes de tratamientos previos, la edad, el sexo o la etnia.
Se describió otro ensayo de fase II con un diseño diferente. En este ensayo, los pacientes con osteosarcoma y metástasis pulmonares se sometieron a resección quirúrgica de todos los nódulos pulmonares y luego recibieron por vía inhalada un factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) como tratamiento adyuvante.[15]
- La tasa de SSC a 12 meses para 42 pacientes evaluables inscritos en este estudio fue del 20 % (IC 95 %, 10–34 %).
Se han estudiado los siguientes fármacos quimioterapéuticos y de terapia dirigida para tratar el osteosarcoma y el sarcoma pleomórfico indiferenciado óseo recidivantes:
- Ifosfamida sola y uroprotección con mesna, o combinada con etopósido. La ifosfamida sola y la uroprotección con mesna, o combinada con etopósido fue eficaz en hasta un tercio de los pacientes con osteosarcoma recidivante que no habían recibido este fármaco.[16,17,18,19,20,21]
- Gemcitabina y docetaxel. En una comparación sin aleatorización de 2 dosis de gemcitabina, combinadas con docetaxel, se indicó que una dosis más alta de gemcitabina (900 mg/m2) se relacionó con una tasa de respuesta superior y una supervivencia más prolongada que la dosis más baja de gemcitabina (675 mg/m2) en casos de osteosarcoma recidivante o resistente al tratamiento.[22][Nivel de evidencia C1] También se notificó la eficacia de la combinación de gemcitabina (en dosis de 900 mg/m2) y docetaxel en algunos estudios en los que se incluyó a pacientes con enfermedad irresecable.[23,24,25]; [26][Nivel de evidencia C3]
- Ciclofosfamida y etopósido. Se ha observado que la ciclofosfamida y el etopósido son un tratamiento eficaz para el osteosarcoma recidivante.[23]
- Sorafenib. El Italian Sarcoma Group notificó respuestas objetivas poco frecuentes y estabilización de la enfermedad cuando se usó sorafenib en pacientes con osteosarcoma recidivante.[27]
- Sorafenib y everólimus. El Italian Sarcoma Group también informó el desenlace de pacientes con osteosarcoma metastásico recidivante tratados con una combinación de sorafenib y everólimus. Se observaron 2 respuestas parciales y 2 respuestas menores en 38 pacientes; 17 de los 38 pacientes no presentaron progresión de la enfermedad 6 meses después de ingresar al estudio, pero los efectos tóxicos fueron mayores que para el grupo de sorafenib en monoterapia.[28][Nivel de evidencia B4]
- Regorafenib. En dos ensayos aleatorizados prospectivos con enmascaramiento doble se evaluó la función del regorafenib en el tratamiento del osteosarcoma metastásico recidivante. En ambos estudios se usó el régimen de tratamiento aprobado de 160 mg al día por vía oral durante 21 días seguido de 7 días sin tratamiento. En el ensayo francés se asignó al azar a los pacientes en una proporción de 2:1 a recibir regorafenib y placebo, y se permitió el cruce de los pacientes asignados al grupo de placebo.[29] En el grupo de regorafenib, 17 de 26 pacientes (65 %; IC unilateral 95 %, 47 %) no exhibieron progresión de la enfermedad a las 8 semanas, en comparación con 0 de 12 pacientes en el grupo de placebo. El grupo Sarcoma Alliance for Research Collaboration (SARC) asignó al azar a pacientes adultos en una proporción de 1:1 a regorafenib y placebo.[30] La mediana de supervivencia sin progresión (SSP) mejoró de forma significativa con regorafenib versus placebo: 3,6 meses (IC 95 %, 2,0–7,6 meses) versus 1,7 meses (IC 95 %, 1,2–1,8 meses), respectivamente (CRI, 0,42; IC 95 %, 0,21–0,85; P = 0,017).
- Dinutuximab en combinación con GM-CSF. El COG realizó un estudio de fase II de dinutuximab, un anticuerpo antidisialogangliósido, en combinación con GM-CSF para pacientes con osteosarcoma recidivante. De 39 pacientes, 28 presentaron un evento y solo el 28,2 % (IC 95 %, 15–44,9 %) no tuvieron complicaciones en el momento del punto de referencia a los 12 meses. El dinutuximab no mostró evidencia suficiente de eficacia.[31]
- Denosumab. El COG llevó a cabo un estudio de fase II de denosumab, un anticuerpo monoclonal totalmente humano del ligando del receptor activador del factor nuclear κ β (RANKL). Los pacientes de 11 a 49 años con osteosarcoma recidivante fueron aptos para participar en el estudio. Los pacientes con enfermedad cuantificable fueron aptos para la cohorte 1 y los pacientes sometidos a resección quirúrgica completa de todos los sitios de enfermedad recidivante fueron aptos para la cohorte 2. Ninguna de las cohortes tuvo una tasa sin complicaciones que excediera la tasa histórica que se obtuvo en ensayos previos del COG para cumplir con los criterios de eficacia definidos para el estudio.[32]
- Lenvatinib con ifosfamida y etopósido. Un grupo multiinstitucional realizó un estudio de fase I/II en el que se usó una combinación de lenvatinib, ifosfamida y etopósido para tratar a pacientes con osteosarcoma recidivante o resistente al tratamiento.[33][Nivel de evidencia B4] En el estudio se indicó la administración de dosis de fase II para cada fármaco: 1) 14 mg/m2 al día de lenvatinib (con una dosis diaria máxima de 24 mg), 2) 100 mg/m2 al día de etopósido y 3) 3000 mg/m2 al día de ifosfamida. Estos fármacos se administraron a los pacientes por vía intravenosa los días 1 a 3 de cada ciclo de 3 semanas, durante 5 ciclos como máximo. En total, 35 pacientes de los ensayos de fase I (cohorte 3A; n = 15) y de fase II (cohorte 3B; n = 20) recibieron las dosis recomendadas en la fase II y sus resultados se agruparon. De los 35 pacientes, 18 presentaron tasas de SSP del 51 % (IC 95 %, 34–69 %) al cabo de 4 meses, según la estimación binomial.
- Cabozantinib. En un ensayo clínico de fase II de este inhibidor de VEGFR2 y MET se evaluó a 43 pacientes de 12 años o más con osteosarcoma en recaída, 39 de los cuales tenían metástasis pulmonares. La tasa de SSP a 6 meses fue del 33 % y 5 pacientes (12 %) presentaron una respuesta parcial.[34]
- Apatinib. En un ensayo clínico de fase II de este inhibidor de VEGFR2 participaron 37 pacientes de 16 años o más. De los pacientes, 16 (43 %) presentaron una respuesta parcial y la tasa de SSP a 4 meses fue del 57 %.[35] En una revisión retrospectiva de 19 pacientes de osteosarcoma tratados con apatinib, 3 pacientes presentaron una respuesta parcial (16 %).[36]
- Anlotinib. En un análisis retrospectivo se notificó la eficacia del anlotinib, un inhibidor multiobjetivo de tirosina–cinasas, en pacientes con osteosarcoma metastásico recidivante.[37] En el estudio se incluyó a 15 pacientes que recibieron tratamiento en China entre junio de 2018 y abril de 2020. La mediana de SSP fue de 9,8 meses (± 0,9 meses). La tasa de SSP a 6 meses fue del 73 %, y la tasa de SSP a 10 meses fue del 33 %. La mediana de SG fue de 11,4 meses (± 0,6 meses). Ningún paciente logró una respuesta completa.
- Inmunoterapia. El osteosarcoma con frecuencia expresa PD-1, pero los ensayos con inhibidores de PD-1 han sido decepcionantes.[38,39,40] En estos tres estudios, se observaron respuestas objetivas en 1 de 14, 0 de 10 y 1 de 19 pacientes. También está en estudio la terapia de células T con receptores de antígeno quimérico (CAR) que expresan HER2.[41]
- Quimioterapia de dosis altas con trasplante de células madre hematopoyéticas (TCMH) autógeno. En un estudio se analizó la adición de dosis altas de quimioterapia con TCMH autógeno para tratar a pacientes coreanos con osteosarcoma en recaída que lograron una respuesta completa con la terapia de rescate.[42] De 25 pacientes que lograron una respuesta completa con terapia de rescate, 15 se asignaron a recibir quimioterapia de dosis altas con TCMH autógeno por elección del investigador. En un análisis de subgrupos de desenlaces en pacientes que lograron una respuesta completa, no hubo diferencias significativas en las tasas de SG a 5 años entre los pacientes que recibieron quimioterapia de dosis altas con TCMH autógeno y los que no la recibieron (83,9 % ± 0,1 % para 13 de 15 pacientes vs. 80,0 % ± 0,1 % para 8 de 10 pacientes, respectivamente; P = 0,923).
Radiofármacos y radioterapia
Es posible que las dosis altas de ácido samario Sm 153-ácido etilendiaminotetrametilenfosfónico (153Sm-EDTMP), junto con el apoyo de células madre periféricas, proporcionen una paliación significativa del dolor en pacientes con metástasis óseas.[43,44,45,46] Los efectos tóxicos del 153Sm-EDTMP son principalmente hematológicos.[47][Nivel de evidencia C2]
En una revisión retrospectiva de una sola institución se notificó que la radioterapia con fracciones de dosis altas (2 Gy/fracción) fue un método de paliación útil para los pacientes con osteosarcoma recidivante.[48][Nivel de evidencia C3] Se administraron 32 cursos de radioterapia paliativa a 20 pacientes con enfermedad primaria metastásica sintomática o recidivante local. De los 32 cursos, 24 (75 %) se relacionaron con mejora de los síntomas. Las dosis más altas de radioterapia se correlacionaron con una mayor duración de la respuesta de los síntomas.
La paliación de las lesiones dolorosas en niños con enfermedad recidivante o progresiva se puede lograr con un curso corto (10 fracciones o menos) de radioterapia. En un estudio retrospectivo de 213 niños con diversas neoplasias malignas que recibieron tratamiento con radioterapia de curso corto, el 85 % de los pacientes lograron alivio completo o parcial del dolor, con niveles bajos de toxicidad.[49]
Opciones de tratamiento para la recidiva local
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo que presentan recidiva local son las siguientes:
- Cirugía para extirpar el tumor.
El desenlace posterior a la recaída de los pacientes con recidiva local es bastante precario [50,51,52] y la supervivencia en los pacientes con recidiva local y metástasis sistémicas, previas o simultáneas, es precaria.[53]
En dos series retrospectivas de una sola institución, se notificó una tasa de supervivencia del 10 % al 40 % después de la recidiva local sin metástasis sistémica relacionada.[53,54,55,56]
En una revisión retrospectiva del Italian Sarcoma Group se identificaron 62 pacientes (mediana de edad, 21 años) con recidivas locales.[57] Al cabo de una mediana de seguimiento de 43 meses (intervalo, 5–235 meses), la tasa de supervivencia posrecaída local a 5 años fue del 37 %, que fue significativamente superior para los pacientes con un intervalo libre de recaída local más largo (≤24 meses, 31 % vs >24 meses, 61,5 %; P = 0,03), ausencia de metástasis a distancia (sin metástasis a distancia, 56 % vs. metástasis a distancia, 11,5 %; P = 0,0001) y logro de la segunda remisión completa (RC) mediante resección quirúrgica (sin segunda RC, 0 % vs. segunda RC, 58,5 %; P = 0,0001). No se encontraron diferencias en la supervivencia posrecaída local según la edad, y no hubo beneficio por la administración de quimioterapia.
La incidencia de recaída local fue más alta en los pacientes con una respuesta patológica deficiente a la quimioterapia en el tumor primario y en los pacientes con márgenes quirúrgicos inadecuados.[50,55]
Opciones de tratamiento para la recidiva pulmonar sola
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo que recidivaron solo en el pulmón son las siguientes:
- Cirugía para extirpar el tumor.
- Quimioterapia o terapia dirigida. Para obtener más información, consultar la sección Quimioterapia y terapia dirigida.
- Radioterapia.
Las resecciones repetidas de recidivas pulmonares pueden llevar a un control prolongado de la enfermedad y, posiblemente, a la curación de algunos pacientes.[13,58] La tasa de supervivencia es inferior al 5 % para los pacientes con enfermedad metastásica irresecable.[6,59] La tasa de SSC a 5 años oscila entre el 20 % y el 45 % para los pacientes que se someten a resección quirúrgica completa de todas las metástasis pulmonares.[4,12,13]; [60][Nivel de evidencia C1]
Los factores relacionados con un mejor desenlace son los siguientes:[4,6,61,62,63]
- Menos nódulos pulmonares.
- Metástasis pulmonares unilaterales.
- Intervalos más prolongados entre la resección del tumor primario y las metástasis.
- Ubicación del tumor en la periferia del pulmón.
Cerca del 50 % de los pacientes con una lesión pulmonar aislada más de 1 año después del diagnóstico sobrevivieron a largo plazo después de una metastasectomía. La quimioterapia no ofreció ninguna ventaja.[64][Nivel de evidencia C1]
Para controlar el osteosarcoma, es necesaria la resección quirúrgica de todos los tumores macroscópicos. No obstante, las recomendaciones con respecto al abordaje quirúrgico para el tratamiento de las metástasis pulmonares de un osteosarcoma son contradictorias. Hay varias opciones disponibles para resecar nódulos pulmonares en pacientes con osteosarcoma, como la toracoscopia y toracotomía con palpación del pulmón atelectásico. Cuando los pacientes tienen nódulos identificados solo en un pulmón, algunos cirujanos abogan por la toracoscopia, mientras que otros lo hacen por la toracotomía unilateral o por la toracotomía bilateral. La toracotomía bilateral se puede realizar como un procedimiento quirúrgico único usando un abordaje por esternotomía mediana o una incisión en concha de almeja, o también se puede hacer como toracotomías bilaterales por etapas.
Evidencia (abordaje quirúrgico de la recidiva pulmonar sola del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo):
- El St. Jude Children's Research Hospital informó sobre 81 pacientes que presentaron nódulos pulmonares unilaterales identificados mediante tomografía computarizada (TC) en el momento la presentación inicial.[65] Se realizó toracotomía unilateral y no se exploró el hemitórax contralateral. En el momento de la toracotomía, 44 de 81 pacientes tenían 1 nódulo solitario; 15 de 81 pacientes tenían 2 nódulos; 16 de 81 pacientes tenían entre 3 y 5 nódulos; y 6 de 81 pacientes presentaban más de 5 nódulos. El resto de los pacientes que no se consideraron aptos para una resección se excluyeron del análisis.
- De los 81 pacientes, 39 presentaron recidiva pulmonar subsiguiente; en la mayoría de los pacientes, la recidiva se presentó en el transcurso de los 6 meses siguientes.
- En los primeros 6 meses, 9 de 81 pacientes presentaron recidiva ipsilateral, y 10 de 81 pacientes presentaron recidiva contralateral. Al cabo de 2 años de la toracotomía inicial, 13 de 81 pacientes presentaron recidiva ipsilateral; 19 de 81 pacientes presentaron recidiva contralateral; y 2 de 81 pacientes presentaron recidiva bilateral.
- La SG fue similar en los pacientes con recidiva ipsilateral o bilateral.
- El Memorial Sloan Kettering Cancer Center informó sobre recidiva de la enfermedad pulmonar metastásica después del tratamiento inicial de un osteosarcoma. De los pacientes, 14 tenían nódulos pulmonares identificados en un solo pulmón mediante TC. Se identificaron 9 pacientes antes de 2 años a partir del momento del diagnóstico inicial (metástasis tempranas) y 5 pacientes se identificaron después de 2 años tras el diagnóstico inicial (metástasis tardías).[62] De 9 pacientes con metástasis tempranas, 7 se sometieron a toracotomías contralaterales por etapas y a 6 de 7 se les extirparon nódulos del pulmón contralateral, a pesar de los resultados negativos en la TC.
- La ausencia de un grupo de comparación impide la evaluación del efecto de la toracotomía contralateral en la SSC o la SG posteriores.
- El mismo grupo amplió el análisis para incluir una revisión retrospectiva de 161 toracotomías realizadas a 88 pacientes de osteosarcoma metastásico en el pulmón.[66] En esta serie ampliada, la TC no logró identificar un tercio de las metástasis pulmonares confirmadas mediante un examen patológico.
El COG está realizando un ensayo aleatorizado (AOST2031 [NCT05235165]) para comparar el efecto de la toracotomía con la cirugía toracoscópica para extirpar metástasis pulmonares. Para obtener más información, consultar la sección Opciones de tratamiento en evaluación clínica.
Es posible que la radioterapia externa proporcione control local de la enfermedad irresecable recidivante, sintomática o metastásica. Las técnicas de radioterapia conocidas como radioterapia ablativa estereotáctica (SABR) o radioterapia corporal estereotáctica (SBRT) permiten administrar dosis elevadas muy conformadas. Este tipo de radioterapia administra el tratamiento con gran conformidad y precisión durante un corto período de tiempo, y proporcionan alivio adecuado y control local.[67]
Opciones de tratamiento para la recidiva con metástasis óseas solas
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo que recidivaron solo en los huesos son las siguientes:
- Cirugía para extirpar el tumor.
- 153Sm-EDTMP, con apoyo de células madre o sin este.
- Quimioterapia o terapia dirigida. Para obtener más información, consultar la sección Quimioterapia y terapia dirigida.
- Radioterapia.
Los pacientes de osteosarcoma que tienen metástasis óseas tienen un pronóstico precario. En una serie grande, la tasa de SSC a 5 años fue del 11 %.[68] Los pacientes con recaída ósea solitaria tardía tienen una tasa de SSC a 5 años de alrededor del 30 %.[68,69,70,71]
En los pacientes con lesiones óseas múltiples irresecables, el uso de 153Sm-EDTMP con apoyo de células madre o sin este a veces estabiliza la enfermedad o alivia el dolor.[47]
Es posible que la radioterapia externa proporcione control local de la enfermedad irresecable recidivante, sintomática o metastásica. Las técnicas de radioterapia conocidas como radioterapia ablativa estereotáctica (SABR) o radioterapia corporal estereotáctica (SBRT) permiten administrar dosis elevadas muy conformadas. Este tipo de radioterapia administra el tratamiento con gran conformidad y precisión durante un corto período de tiempo, y proporcionan alivio adecuado y control local.[67]
Opciones de tratamiento para la segunda recidiva del osteosarcoma
Las opciones de tratamiento para los pacientes con osteosarcoma o sarcoma pleomórfico indiferenciado (SPI) óseo que recidivaron dos veces son las siguientes:
- Cirugía para extirpar el tumor o quimioterapia.
- Quimioterapia o terapia dirigida. Para obtener más información, consultar la sección Quimioterapia y terapia dirigida.
Evidencia (cirugía o quimioterapia):
- El Cooperative German-Austrian-Swiss Osteosarcoma Study Group informó sobre 249 pacientes que presentaron una segunda recidiva del osteosarcoma. El tratamiento principal fue la repetición de la resección quirúrgica de la enfermedad recidivante.[72]
- De estos pacientes, 197 murieron y 37 sobrevivieron con RC (24 luego de la tercera respuesta completa, y 13 después de la cuarta respuesta o subsiguientes respuestas completas).
- De los pacientes que no obtuvieron la remisión quirúrgica, 15 permanecieron vivos, pero el seguimiento de estos pacientes fue muy breve.
- El Spanish Group for Research on Sarcoma notificó los resultados de un ensayo de fase II de pacientes con osteosarcoma recidivante o resistente al tratamiento que recibieron gemcitabina y sirólimus.[73][Nivel de evidencia C3]
- La tasa de SSP a los 4 meses fue del 44 %.
- Luego de una revisión radiológica central de 33 pacientes evaluables, se reportaron 2 respuestas parciales y 14 estabilizaciones de la enfermedad (48,5 %).
- El COG notificó los desenlaces de los pacientes con osteosarcoma recidivante de 7 ensayos de fase II; en ningún caso se encontró beneficio del tratamiento.[15]
- La tasa de SSC para 96 pacientes con osteosarcoma y enfermedad cuantificable fue del 12 % a los 4 meses (IC 95 %, 6–19 %).
- No hubo ninguna diferencia significativa en la SSC entre los ensayos, de acuerdo con el número de regímenes de tratamientos previos, la edad, el sexo o la etnia.
- Se describió otro ensayo de fase II con un diseño diferente. En este ensayo, los pacientes con osteosarcoma y metástasis pulmonares se sometieron a resección quirúrgica de todos los nódulos pulmonares, y luego recibieron por vía inhalada GM-CSF como tratamiento adyuvante.[15]
- La tasa de SSC a 12 meses para 42 pacientes evaluables inscritos en este estudio fue del 20 % (IC 95 %, 10–34 %).
Opciones de tratamiento en evaluación clínica
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presentan ejemplos de ensayos clínicos nacionales o institucionales en curso:
- GD2-CAR PERSIST (NCT04539366) (Testing a New Immune Cell Therapy, GD2-Targeted Modified T-cells (GD2CART), in Children, Adolescents, and Young Adults with Relapsed/Refractory Osteosarcoma and Neuroblastoma): se trata de un ensayo de fase I para determinar los efectos secundarios y la mejor dosis de GD2CART con el fin de que sea eficaz contra las células tumorales positivas para GD2.
- AOST2031 (NCT05235165) (Thoracotomy Versus Thoracoscopic Management of Pulmonary Metastases in Patients With Osteosarcoma): en este ensayo de fase III se compara el efecto de la toracotomía con la cirugía toracoscópica (cirugía toracoscópica asistida por video) en el tratamiento de pacientes con osteosarcoma que se extendió al pulmón (metástasis pulmonar). Este ensayo se realiza para evaluar los 2 métodos quirúrgicos diferentes para estos pacientes y determinar qué procedimiento es mejor.
- NCT03811886 (Natalizumab for the Treatment of Recurrent, Refractory, or Progressive Pulmonary Metastatic Osteosarcoma): este es un ensayo de fase I/II en el que se evalúa la inocuidad y la respuesta al natalizumab en pacientes con metástasis pulmonares que progresaron, recayeron o se volvieron resistentes al tratamiento sistémico.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
- Gelderblom H, Jinks RC, Sydes M, et al.: Survival after recurrent osteosarcoma: data from 3 European Osteosarcoma Intergroup (EOI) randomized controlled trials. Eur J Cancer 47 (6): 895-902, 2011.
- Hauben EI, Bielack S, Grimer R, et al.: Clinico-histologic parameters of osteosarcoma patients with late relapse. Eur J Cancer 42 (4): 460-6, 2006.
- Ferrari S, Briccoli A, Mercuri M, et al.: Late relapse in osteosarcoma. J Pediatr Hematol Oncol 28 (7): 418-22, 2006.
- Kempf-Bielack B, Bielack SS, Jürgens H, et al.: Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 23 (3): 559-68, 2005.
- Spraker-Perlman HL, Barkauskas DA, Krailo MD, et al.: Factors influencing survival after recurrence in osteosarcoma: A report from the Children's Oncology Group. Pediatr Blood Cancer 66 (1): e27444, 2019.
- Bacci G, Briccoli A, Longhi A, et al.: Treatment and outcome of recurrent osteosarcoma: experience at Rizzoli in 235 patients initially treated with neoadjuvant chemotherapy. Acta Oncol 44 (7): 748-55, 2005.
- Rejin K, Aykan OA, Omer G, et al.: Intra-abdominal metastasis in osteosarcoma: survey and literature review. Pediatr Hematol Oncol 28 (7): 609-15, 2011.
- Goorin AM, Shuster JJ, Baker A, et al.: Changing pattern of pulmonary metastases with adjuvant chemotherapy in patients with osteosarcoma: results from the multiinstitutional osteosarcoma study. J Clin Oncol 9 (4): 600-5, 1991.
- Harting MT, Blakely ML: Management of osteosarcoma pulmonary metastases. Semin Pediatr Surg 15 (1): 25-9, 2006.
- Pastorino U, Gasparini M, Tavecchio L, et al.: The contribution of salvage surgery to the management of childhood osteosarcoma. J Clin Oncol 9 (8): 1357-62, 1991.
- Skinner KA, Eilber FR, Holmes EC, et al.: Surgical treatment and chemotherapy for pulmonary metastases from osteosarcoma. Arch Surg 127 (9): 1065-70; discussion 1070-1, 1992.
- Chou AJ, Merola PR, Wexler LH, et al.: Treatment of osteosarcoma at first recurrence after contemporary therapy: the Memorial Sloan-Kettering Cancer Center experience. Cancer 104 (10): 2214-21, 2005.
- Harting MT, Blakely ML, Jaffe N, et al.: Long-term survival after aggressive resection of pulmonary metastases among children and adolescents with osteosarcoma. J Pediatr Surg 41 (1): 194-9, 2006.
- Lautz TB, Farooqui Z, Jenkins T, et al.: Thoracoscopy vs thoracotomy for the management of metastatic osteosarcoma: A Pediatric Surgical Oncology Research Collaborative Study. Int J Cancer 148 (5): 1164-1171, 2021.
- Lagmay JP, Krailo MD, Dang H, et al.: Outcome of Patients With Recurrent Osteosarcoma Enrolled in Seven Phase II Trials Through Children's Cancer Group, Pediatric Oncology Group, and Children's Oncology Group: Learning From the Past to Move Forward. J Clin Oncol 34 (25): 3031-8, 2016.
- Harris MB, Cantor AB, Goorin AM, et al.: Treatment of osteosarcoma with ifosfamide: comparison of response in pediatric patients with recurrent disease versus patients previously untreated: a Pediatric Oncology Group study. Med Pediatr Oncol 24 (2): 87-92, 1995.
- Miser JS, Kinsella TJ, Triche TJ, et al.: Ifosfamide with mesna uroprotection and etoposide: an effective regimen in the treatment of recurrent sarcomas and other tumors of children and young adults. J Clin Oncol 5 (8): 1191-8, 1987.
- Kung FH, Pratt CB, Vega RA, et al.: Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A Pediatric Oncology Group Phase II study. Cancer 71 (5): 1898-903, 1993.
- Berrak SG, Pearson M, Berberoğlu S, et al.: High-dose ifosfamide in relapsed pediatric osteosarcoma: therapeutic effects and renal toxicity. Pediatr Blood Cancer 44 (3): 215-9, 2005.
- Palmerini E, Setola E, Grignani G, et al.: High Dose Ifosfamide in Relapsed and Unresectable High-Grade Osteosarcoma Patients: A Retrospective Series. Cells 9 (11): , 2020.
- Tirtei E, Campello A, Sciannameo V, et al.: Prolonged 14-day continuous infusion of high-dose ifosfamide for patients with relapsed and refractory high-grade osteosarcoma: a retrospective multicentre cohort study. BMC Cancer 24 (1): 747, 2024.
- Lee JA, Jeon DG, Cho WH, et al.: Higher Gemcitabine Dose Was Associated With Better Outcome of Osteosarcoma Patients Receiving Gemcitabine-Docetaxel Chemotherapy. Pediatr Blood Cancer 63 (9): 1552-6, 2016.
- Massimo B, Giovanni G, Stefano F, et al.: Phase 2 trial of two courses of cyclophosphamide and etoposide for relapsed high-risk osteosarcoma patients. Cancer 115 (13): 2980-7, 2009.
- Navid F, Willert JR, McCarville MB, et al.: Combination of gemcitabine and docetaxel in the treatment of children and young adults with refractory bone sarcoma. Cancer 113 (2): 419-25, 2008.
- Qi WX, He AN, Tang LN, et al.: Efficacy and safety of gemcitabine-docetaxel combination therapy for recurrent or refractory high-grade osteosarcoma in China: a retrospective study of 18 patients. Jpn J Clin Oncol 42 (5): 427-31, 2012.
- Palmerini E, Jones RL, Marchesi E, et al.: Gemcitabine and docetaxel in relapsed and unresectable high-grade osteosarcoma and spindle cell sarcoma of bone. BMC Cancer 16: 280, 2016.
- Grignani G, Palmerini E, Dileo P, et al.: A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. Ann Oncol 23 (2): 508-16, 2012.
- Grignani G, Palmerini E, Ferraresi V, et al.: Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. Lancet Oncol 16 (1): 98-107, 2015.
- Duffaud F, Mir O, Boudou-Rouquette P, et al.: Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol 20 (1): 120-133, 2019.
- Davis LE, Bolejack V, Ryan CW, et al.: Randomized Double-Blind Phase II Study of Regorafenib in Patients With Metastatic Osteosarcoma. J Clin Oncol 37 (16): 1424-1431, 2019.
- Hingorani P, Krailo M, Buxton A, et al.: Phase 2 study of anti-disialoganglioside antibody, dinutuximab, in combination with GM-CSF in patients with recurrent osteosarcoma: A report from the Children's Oncology Group. Eur J Cancer 172: 264-275, 2022.
- Janeway KA, Chou AJ, Hingorani P, et al.: AOST1321, a phase 2 trial of RANKL antibody, denosumab, in 2 cohorts of patients with recurrent or refractory osteosarcoma: a report from the Children's Oncology Group. [Abstract] 2019 Connective Tissue Oncology Society (CTOS) Annual Meeting, November 13-16, 2019, Tokyo, Japan. A-3254462, 2019. Available online. Last accessed March 30, 2023.
- Gaspar N, Venkatramani R, Hecker-Nolting S, et al.: Lenvatinib with etoposide plus ifosfamide in patients with refractory or relapsed osteosarcoma (ITCC-050): a multicentre, open-label, multicohort, phase 1/2 study. Lancet Oncol 22 (9): 1312-1321, 2021.
- Italiano A, Mir O, Mathoulin-Pelissier S, et al.: Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): a multicentre, single-arm, phase 2 trial. Lancet Oncol 21 (3): 446-455, 2020.
- Xie L, Xu J, Sun X, et al.: Apatinib for Advanced Osteosarcoma after Failure of Standard Multimodal Therapy: An Open Label Phase II Clinical Trial. Oncologist 24 (7): e542-e550, 2019.
- Tian Z, Liu H, Zhang F, et al.: Retrospective review of the activity and safety of apatinib and anlotinib in patients with advanced osteosarcoma and soft tissue sarcoma. Invest New Drugs 38 (5): 1559-1569, 2020.
- Li H, Li Y, Song L, et al.: Retrospective review of safety and efficacy of anlotinib in advanced osteosarcoma with metastases after failure of standard multimodal therapy. Asia Pac J Clin Oncol 19 (5): e314-e319, 2023.
- Tawbi HA, Burgess M, Bolejack V, et al.: Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol 18 (11): 1493-1501, 2017.
- Le Cesne A, Marec-Berard P, Blay JY, et al.: Programmed cell death 1 (PD-1) targeting in patients with advanced osteosarcomas: results from the PEMBROSARC study. Eur J Cancer 119: 151-157, 2019.
- Boye K, Longhi A, Guren T, et al.: Pembrolizumab in advanced osteosarcoma: results of a single-arm, open-label, phase 2 trial. Cancer Immunol Immunother 70 (9): 2617-2624, 2021.
- Ahmed N, Brawley VS, Hegde M, et al.: Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol 33 (15): 1688-96, 2015.
- Kang SH, Kim W, Lee JS, et al.: High-dose chemotherapy followed by autologous stem cell transplantation in pediatric patients with relapsed osteosarcoma. Pediatr Blood Cancer 70 (4): e30233, 2023.
- Anderson PM, Wiseman GA, Dispenzieri A, et al.: High-dose samarium-153 ethylene diamine tetramethylene phosphonate: low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. J Clin Oncol 20 (1): 189-96, 2002.
- Franzius C, Bielack S, Flege S, et al.: High-activity samarium-153-EDTMP therapy followed by autologous peripheral blood stem cell support in unresectable osteosarcoma. Nuklearmedizin 40 (6): 215-20, 2001.
- Sauerbrey A, Bielack S, Kempf-Bielack B, et al.: High-dose chemotherapy (HDC) and autologous hematopoietic stem cell transplantation (ASCT) as salvage therapy for relapsed osteosarcoma. Bone Marrow Transplant 27 (9): 933-7, 2001.
- Fagioli F, Aglietta M, Tienghi A, et al.: High-dose chemotherapy in the treatment of relapsed osteosarcoma: an Italian sarcoma group study. J Clin Oncol 20 (8): 2150-6, 2002.
- Loeb DM, Garrett-Mayer E, Hobbs RF, et al.: Dose-finding study of 153Sm-EDTMP in patients with poor-prognosis osteosarcoma. Cancer 115 (11): 2514-22, 2009.
- Chen EL, Yoo CH, Gutkin PM, et al.: Outcomes for pediatric patients with osteosarcoma treated with palliative radiotherapy. Pediatr Blood Cancer 67 (1): e27967, 2020.
- Sudmeier LJ, Madden N, Zhang C, et al.: Palliative radiotherapy for children: Symptom response and treatment-associated toxicity according to radiation therapy dose and fractionation. Pediatr Blood Cancer 70 (4): e30195, 2023.
- Weeden S, Grimer RJ, Cannon SR, et al.: The effect of local recurrence on survival in resected osteosarcoma. Eur J Cancer 37 (1): 39-46, 2001.
- Bacci G, Ferrari S, Lari S, et al.: Osteosarcoma of the limb. Amputation or limb salvage in patients treated by neoadjuvant chemotherapy. J Bone Joint Surg Br 84 (1): 88-92, 2002.
- Rodriguez-Galindo C, Shah N, McCarville MB, et al.: Outcome after local recurrence of osteosarcoma: the St. Jude Children's Research Hospital experience (1970-2000). Cancer 100 (9): 1928-35, 2004.
- Bacci G, Longhi A, Cesari M, et al.: Influence of local recurrence on survival in patients with extremity osteosarcoma treated with neoadjuvant chemotherapy: the experience of a single institution with 44 patients. Cancer 106 (12): 2701-6, 2006.
- Grimer RJ, Sommerville S, Warnock D, et al.: Management and outcome after local recurrence of osteosarcoma. Eur J Cancer 41 (4): 578-83, 2005.
- Bacci G, Forni C, Longhi A, et al.: Local recurrence and local control of non-metastatic osteosarcoma of the extremities: a 27-year experience in a single institution. J Surg Oncol 96 (2): 118-23, 2007.
- Nathan SS, Gorlick R, Bukata S, et al.: Treatment algorithm for locally recurrent osteosarcoma based on local disease-free interval and the presence of lung metastasis. Cancer 107 (7): 1607-16, 2006.
- Palmerini E, Torricelli E, Cascinu S, et al.: Is there a role for chemotherapy after local relapse in high-grade osteosarcoma? Pediatr Blood Cancer 66 (8): e27792, 2019.
- Briccoli A, Rocca M, Salone M, et al.: Resection of recurrent pulmonary metastases in patients with osteosarcoma. Cancer 104 (8): 1721-5, 2005.
- Tabone MD, Kalifa C, Rodary C, et al.: Osteosarcoma recurrences in pediatric patients previously treated with intensive chemotherapy. J Clin Oncol 12 (12): 2614-20, 1994.
- Briccoli A, Rocca M, Salone M, et al.: High grade osteosarcoma of the extremities metastatic to the lung: long-term results in 323 patients treated combining surgery and chemotherapy, 1985-2005. Surg Oncol 19 (4): 193-9, 2010.
- Aljubran AH, Griffin A, Pintilie M, et al.: Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. Ann Oncol 20 (6): 1136-41, 2009.
- Su WT, Chewning J, Abramson S, et al.: Surgical management and outcome of osteosarcoma patients with unilateral pulmonary metastases. J Pediatr Surg 39 (3): 418-23; discussion 418-23, 2004.
- Letourneau PA, Xiao L, Harting MT, et al.: Location of pulmonary metastasis in pediatric osteosarcoma is predictive of outcome. J Pediatr Surg 46 (7): 1333-7, 2011.
- Daw NC, Chou AJ, Jaffe N, et al.: Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br J Cancer 112 (2): 278-82, 2015.
- Karplus G, McCarville MB, Smeltzer MP, et al.: Should contralateral exploratory thoracotomy be advocated for children with osteosarcoma and early unilateral pulmonary metastases? J Pediatr Surg 44 (4): 665-71, 2009.
- Heaton TE, Hammond WJ, Farber BA, et al.: A 20-year retrospective analysis of CT-based pre-operative identification of pulmonary metastases in patients with osteosarcoma: A single-center review. J Pediatr Surg 52 (1): 115-119, 2017.
- Brown LC, Lester RA, Grams MP, et al.: Stereotactic body radiotherapy for metastatic and recurrent ewing sarcoma and osteosarcoma. Sarcoma 2014: 418270, 2014.
- Bacci G, Longhi A, Bertoni F, et al.: Bone metastases in osteosarcoma patients treated with neoadjuvant or adjuvant chemotherapy: the Rizzoli experience in 52 patients. Acta Orthop 77 (6): 938-43, 2006.
- Aung L, Gorlick R, Healey JH, et al.: Metachronous skeletal osteosarcoma in patients treated with adjuvant and neoadjuvant chemotherapy for nonmetastatic osteosarcoma. J Clin Oncol 21 (2): 342-8, 2003.
- Jaffe N, Pearson P, Yasko AW, et al.: Single and multiple metachronous osteosarcoma tumors after therapy. Cancer 98 (11): 2457-66, 2003.
- Franke M, Hardes J, Helmke K, et al.: Solitary skeletal osteosarcoma recurrence. Findings from the Cooperative Osteosarcoma Study Group. Pediatr Blood Cancer 56 (5): 771-6, 2011.
- Bielack SS, Kempf-Bielack B, Branscheid D, et al.: Second and subsequent recurrences of osteosarcoma: presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol 27 (4): 557-65, 2009.
- Martin-Broto J, Redondo A, Valverde C, et al.: Gemcitabine plus sirolimus for relapsed and progressing osteosarcoma patients after standard chemotherapy: a multicenter, single-arm phase II trial of Spanish Group for Research on Sarcoma (GEIS). Ann Oncol 28 (12): 2994-2999, 2017.
Actualizaciones más recientes a este resumen (02 / 20 / 2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se añadió Efecto del paso del tiempo sobre los factores pronósticos como subsección nueva.
Se añadió a Bielack et al. como referencia 54.
Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo localizados
Se añadió texto sobre los resultados de un ensayo del Sarcoma Group francés en el que se suprimieron las dosis altas de metotrexato en los pacientes adultos con osteosarcoma (se citó a Blay et al. como referencia 35).
Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo recidivantes
Se añadió a Tirtei et al. como referencia 21.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo son:
- Holcombe Edwin Grier, MD
- Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children's Cancer Center and Blood Disorders Harvard Medical School)
- William H. Meyer, MD
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Nita Louise Seibel, MD (National Cancer Institute)
- Malcolm A. Smith, MD, PhD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento del osteosarcoma y el sarcoma pleomórfico indiferenciado óseo. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en: https://www.cancer.gov/espanol/tipos/hueso/pro/tratamiento-osteosarcoma-pdq. Fecha de acceso: <MM/DD/YYYY>.
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.
Última revisión: 2025-02-20

