Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Incidencia
El estesioneuroblastoma (también llamado neuroblastoma olfatorio u olfativo) es un tumor muy raro de células redondas y pequeñas que surge del neuroepitelio nasal. Menos del 10 % de los casos se presentan durante la niñez y la adolescencia.[1,2] La incidencia anual del estesioneuroblastoma es de 0,1 casos por 100 000 niños menores de 15 años.[3] En la población pediátrica, la mediana de edad de los casos es de 10 años, sin predilección por sexo o raza.[2]
A pesar de su escasa frecuencia, el estesioneuroblastoma es el cáncer de cavidad nasal más común en pacientes pediátricos y representó el 28 % de los casos en un estudio del Surveillance, Epidemiology, and End Results (SEER) Program.[1]
Referencias:
- Benoit MM, Bhattacharyya N, Faquin W, et al.: Cancer of the nasal cavity in the pediatric population. Pediatrics 121 (1): e141-5, 2008.
- Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
- Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
Característica anatómicas
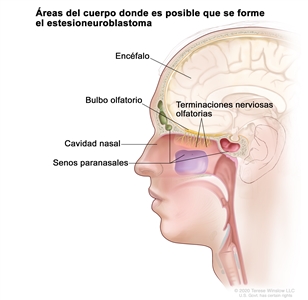
En la Figura 1 se observan las áreas del cuerpo donde es posible que se forme el estesioneuroblastoma, como las terminaciones nerviosas olfatorias, el bulbo olfatorio, la cavidad nasal (fosas nasales), los senos paranasales y el encéfalo.
Cuadro clínico inicial
En la mayoría de los niños, es posible que los primeros síntomas de estesioneuroblastoma sean los siguientes:[1]
- Obstrucción nasal.
- Epistaxis.
- Hiposmia.
- Exoftalmía (exoftalmos).
- Cefaleas.
- Masa nasofaríngea, que se puede extender a nivel local hacia las órbitas, los senos paranasales o el lóbulo frontal.
Referencias:
- Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
Características histológicas y moleculares
Desde el punto de vista histológico, el estesioneuroblastoma en ocasiones se confunde con otros tumores de células redondas y pequeñas de la cavidad nasal, como el carcinoma nasosinusal indiferenciado, el carcinoma de células pequeñas, el melanoma y el rabdomiosarcoma. El estesioneuroblastoma, por lo general, muestra una tinción difusa con enolasa neuroespecífica, sinaptofisina y cromograninas, con una expresión variable de las citoqueratinas.[1]
En 9 centros médicos, se obtuvieron 66 muestras de neuroblastoma olfatorio y de otros tumores cancerosos, entre ellos rabdomiosarcoma alveolar y adenocarcinoma nasosinusal. Estas muestras tumorales se analizaron mediante perfil de metilación del DNA de todo el genoma, análisis del número de copias, inmunohistoquímica y secuenciación de nueva generación. En el análisis de agrupamiento jerárquico no supervisado de metilación del DNA se identificó de forma clara los siguientes 4 grupos:[2]
- El grupo más grande, que abarcó el 64 % de las muestras, presentó características histológicas clásicas de neuroblastoma olfatorio. El 10 % de los casos exhibió variantes recurrentes de DNMT3A y TP53.
- Un segundo grupo estaba formado por 7 casos con fenotipo hipermetilador y variantes de IDH2 que se agruparon con el grupo de carcinomas nasosinusales con variantes de IDH2.
- Un tercer grupo pequeño se caracterizó por presentar hipermetilación sin las variantes de IDH2. Este resultado indica que este grupo podría representar un subgrupo de neuroblastomas olfatorios o una entidad tumoral nasosinusal sin definir.
- El cuarto grupo representó un conjunto heterogéneo de 13 tumores que se agruparon con otras entidades como el adenocarcinoma nasosinusal, el carcinoma nasosinusal de células escamosas, el carcinoma neuroendocrino nasosinusal y el carcinoma nasosinusal indiferenciado.
Con esta información, los autores crearon un algoritmo que incorpora el análisis de metilación para mejorar la exactitud del diagnóstico de esta entidad.[2]
Referencias:
- Su SY, Bell D, Hanna EY: Esthesioneuroblastoma, neuroendocrine carcinoma, and sinonasal undifferentiated carcinoma: differentiation in diagnosis and treatment. Int Arch Otorhinolaryngol 18 (Suppl 2): S149-56, 2014.
- Capper D, Engel NW, Stichel D, et al.: DNA methylation-based reclassification of olfactory neuroblastoma. Acta Neuropathol 136 (2): 255-271, 2018.
Factores pronósticos
En la revisión de una serie de casos múltiples de pacientes en su mayoría adultos, se indica que es posible que los siguientes factores se correlacionen con un pronóstico adverso:[1,2,3]
- Grado histopatológico más alto.
- Márgenes quirúrgicos afectados.
- Metástasis en los ganglios linfáticos cervicales.
Referencias:
- Dulguerov P, Allal AS, Calcaterra TC: Esthesioneuroblastoma: a meta-analysis and review. Lancet Oncol 2 (11): 683-90, 2001.
- Patel SG, Singh B, Stambuk HE, et al.: Craniofacial surgery for esthesioneuroblastoma: report of an international collaborative study. J Neurol Surg B Skull Base 73 (3): 208-20, 2012.
- Herr MW, Sethi RK, Meier JC, et al.: Esthesioneuroblastoma: an update on the massachusetts eye and ear infirmary and massachusetts general hospital experience with craniofacial resection, proton beam radiation, and chemotherapy. J Neurol Surg B Skull Base 75 (1): 58-64, 2014.
Información sobre los estadios del estesioneuroblastoma infantil
Los tumores se estadifican según el sistema de Kadish (consultar el Cuadro 1). En correlación con el estadio de Kadish, las tasas de supervivencia oscilan entre el 90 % (estadio A) y menos del 40 % (estadio D). La mayoría de los pacientes presentan al inicio una enfermedad en estadio localmente avanzado (estadio B y C de Kadish). Los informes de enfermedad metastásica (estadio D de Kadish) varían según los estudios y se describe en tasas del 20 % al 30 %.[1,2,3,4,5,6]
En informes se indica que la tomografía por emisión de positrones combinada con tomografía computarizada (TEP-TC) quizás ayude a estadificar la enfermedad.[7]
| Estadio | Descripción |
|---|---|
| A | Tumor confinado en la cavidad nasal. |
| B | Tumor extendido a los senos paranasales. |
| C | Tumor extendido a los senos paranasales y más allá. |
| D | Presencia de metástasis tumorales. |
Referencias:
- Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
- Benoit MM, Bhattacharyya N, Faquin W, et al.: Cancer of the nasal cavity in the pediatric population. Pediatrics 121 (1): e141-5, 2008.
- Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
- Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
- Dumont B, Fresneau B, Claude L, et al.: Pattern of loco-regional relapses and treatment in pediatric esthesioneuroblastoma: The French very rare tumors group (Fracture) contribution. Pediatr Blood Cancer 67 (4): e28154, 2020.
- Safi C, Spielman D, Otten M, et al.: Treatment Strategies and Outcomes of Pediatric Esthesioneuroblastoma: A Systematic Review. Front Oncol 10: 1247, 2020.
- Broski SM, Hunt CH, Johnson GB, et al.: The added value of 18F-FDG PET/CT for evaluation of patients with esthesioneuroblastoma. J Nucl Med 53 (8): 1200-6, 2012.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es raro, aunque desde 1975 se ha observado un aumento gradual de la incidencia general.[1] Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes especialistas en pediatría y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
- Médicos de atención primaria.
- Cirujanos pediatras.
- Patólogos.
- Radioncólogo pediatras.
- Oncólogos y hematólogos pediatras.
- Oftalmólogos.
- Especialistas en rehabilitación.
- Enfermeros de oncología pediátrica.
- Trabajadores o asistentes sociales.
- Profesionales de la vida infantil.
- Psicólogos.
- Nutricionistas y dietistas.
Para obtener información específica sobre los cuidados médicos de apoyo para niños y adolescentes con cáncer, consultar los resúmenes de Cuidados médicos de apoyo y cuidados paliativos.
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes con cáncer infantil.[2] En estos centros, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. En otros tipos de ensayos clínicos se prueban tratamientos nuevos cuando no hay un tratamiento estándar para el cáncer que se ha diagnosticado. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
Se han logrado mejoras notables en la supervivencia de niños y adolescentes con cáncer. Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más del 50 %.[3,4,5] Los sobrevivientes del cáncer que se presenta en edad pediátrica necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan o se presenten meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los sobrevivientes de cáncer infantil, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
El cáncer infantil (cáncer en edad pediátrica) es una enfermedad rara con cerca de 15 000 casos anuales diagnosticados antes de los 20 años de edad en los Estados Unidos.[6] En la Rare Diseases Act of 2002 de los Estados Unidos se define una enfermedad rara como la que afecta a poblaciones de menos de 200 000 personas. Por lo tanto, todos los cánceres infantiles se consideran enfermedades raras.
La designación de un tumor raro es diferente entre los grupos pediátricos y de adultos. En el caso de los adultos, se considera que un cáncer es raro cuando su incidencia anual es inferior a 6 casos por 100 000 personas. Representan hasta el 24 % de los cánceres diagnosticados en la Unión Europea y alrededor del 20 % de los cánceres diagnosticados en los Estados Unidos.[7,8] Además, tal como se indica a continuación, la designación de un tumor raro durante la niñez y adolescencia no es uniforme entre los grupos internacionales:
- En una iniciativa conjunta de la European Union Joint Action on Rare Cancers y el European Cooperative Study Group for Rare Pediatric Cancers se estimó que el 11 % de todos los cánceres en pacientes menores de 20 años se podrían clasificar como muy raros. Este grupo de consenso definió los cánceres muy raros como los cánceres con incidencia anual inferior a 2 casos por millón de personas. Sin embargo, también se incluyen en este grupo de tumores muy raros otros 3 tipos histológicos (carcinoma de tiroides, melanoma y cáncer de testículo) con incidencias superiores a 2 casos por millón de personas, porque se cuenta con poco conocimiento y experiencia sobre el tratamiento de estos tumores.[9]
- El Children's Oncology Group (COG) define los cánceres raros en pediatría según la lista del subgrupo XI de la International Classification of Childhood Cancer, en la que se incluyen los cánceres de tiroides, los cánceres de piel melanoma y no melanoma, además de los múltiples tipos de carcinomas (por ejemplo, los carcinomas de corteza suprarrenal, los carcinomas de nasofaringe y la mayoría de los carcinomas de tipo adulto, como los cánceres de mama, los cánceres colorrectales, etc.).[10] Estos cánceres representan casi el 5 % de aquellos diagnosticados entre los 0 y 14 años de edad y casi el 27 % de los que se diagnostican entre los 15 a 19 años de edad.[4]
La mayoría de los cánceres del subgrupo XI son melanomas o cánceres de tiroides, mientras que otros tipos de cáncer solo representan el 2 % de los cánceres diagnosticados entre los 0 y 14 años de edad y el 9,3 % de los cánceres entre los 15 y 19 años de edad.
Estudiar estos cánceres raros es un reto por el número bajo de pacientes con cualquier diagnóstico individual, el predominio de estos cánceres raros en adolescentes y la carencia de ensayos clínicos con adolescentes que tienen estos cánceres.
Referencias:
- Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
- American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed February 25, 2025.
- Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
- Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed December 30, 2024.
- Ward E, DeSantis C, Robbins A, et al.: Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 64 (2): 83-103, 2014 Mar-Apr.
- Gatta G, Capocaccia R, Botta L, et al.: Burden and centralised treatment in Europe of rare tumours: results of RARECAREnet-a population-based study. Lancet Oncol 18 (8): 1022-1039, 2017.
- DeSantis CE, Kramer JL, Jemal A: The burden of rare cancers in the United States. CA Cancer J Clin 67 (4): 261-272, 2017.
- Ferrari A, Brecht IB, Gatta G, et al.: Defining and listing very rare cancers of paediatric age: consensus of the Joint Action on Rare Cancers in cooperation with the European Cooperative Study Group for Pediatric Rare Tumors. Eur J Cancer 110: 120-126, 2019.
- Pappo AS, Krailo M, Chen Z, et al.: Infrequent tumor initiative of the Children's Oncology Group: initial lessons learned and their impact on future plans. J Clin Oncol 28 (33): 5011-6, 2010.
Tratamiento del estesioneuroblastoma infantil
El uso de terapia multimodal optimiza las probabilidades de supervivencia: se espera que más del 70 % de los niños sobrevivan 5 años o más después del diagnóstico inicial.[1,2,3,4,5] La progresión neuromeníngea es el tipo más común de manifestación de un fracaso del tratamiento.[5,6][Nivel de evidencia C1]
Las opciones de tratamiento según el estadio de Kadish son las siguientes:[7]
- Estadio A de Kadish: cirugía sola con márgenes limpios. La radioterapia adyuvante se indica para pacientes con márgenes estrechos afectados o con enfermedad residual.
- Estadio B de Kadish: cirugía seguida de radioterapia adyuvante. La función de la quimioterapia adyuvante es controvertida.
- Estadio C de Kadish: abordaje neoadyuvante con quimioterapia, radioterapia o quimiorradiación simultánea, seguido de cirugía.
- Estadio D de Kadish: quimioterapia sistémica y radioterapia dirigida a sitios locales y metastásicos.
Los pilares del tratamiento son la cirugía y la radioterapia. Sin embargo, el estesioneuroblastoma es una neoplasia quimiosensible y el empleo de quimioterapia neoadyuvante quizás facilite la extirpación.[5,7,8,9] La cirugía endoscópica de senos paranasales produce desenlaces en el corto plazo que son similares a los que se obtienen con la resección craneofacial abierta.[10]; [11][Nivel de evidencia C2] Es posible que otras técnicas, como la radiocirugía estereotáctica y la terapia con haz de protones (radioterapia con partículas cargadas), también cumplan una función en el tratamiento de este tumor.[3,12,13]
La disección rutinaria del cuello y la exploración ganglionar no están indicadas si no hay manifestaciones clínicas o radiológicas de enfermedad.[14] El tratamiento de las metástasis en los ganglios linfáticos cervicales se consideró en un artículo de revisión.[14]
En los informes se han indicado resultados prometedores del uso creciente de la resección, y la quimioterapia neoadyuvante o adyuvante para pacientes con enfermedad en estadio avanzado.[2,5,15,16,17]; [18][Nivel de evidencia C1] Se han utilizado regímenes de quimioterapia eficaces que incluyen los siguientes:
- Cisplatino y etopósido, con ifosfamida o sin esta.[19,20]
- Vincristina, actinomicina D y ciclofosfamida con doxorrubicina o sin esta.
- Ifosfamida con etopósido.
- Cisplatino con etopósido o doxorrubicina.[2]
- Vincristina, doxorrubicina y ciclofosfamida.[21]
- Irinotecán con docetaxel.[22][Nivel de evidencia C1]
Referencias:
- Bisogno G, Soloni P, Conte M, et al.: Esthesioneuroblastoma in pediatric and adolescent age. A report from the TREP project in cooperation with the Italian Neuroblastoma and Soft Tissue Sarcoma Committees. BMC Cancer 12: 117, 2012.
- Eich HT, Müller RP, Micke O, et al.: Esthesioneuroblastoma in childhood and adolescence. Better prognosis with multimodal treatment? Strahlenther Onkol 181 (6): 378-84, 2005.
- Lucas JT, Ladra MM, MacDonald SM, et al.: Proton therapy for pediatric and adolescent esthesioneuroblastoma. Pediatr Blood Cancer 62 (9): 1523-8, 2015.
- Berger MH, Lehrich BM, Yasaka TM, et al.: Characteristics and overall survival in pediatric versus adult esthesioneuroblastoma: A population-based study. Int J Pediatr Otorhinolaryngol 144: 110696, 2021.
- Venkatramani R, Pan H, Furman WL, et al.: Multimodality Treatment of Pediatric Esthesioneuroblastoma. Pediatr Blood Cancer 63 (3): 465-70, 2016.
- Dumont B, Fresneau B, Claude L, et al.: Pattern of loco-regional relapses and treatment in pediatric esthesioneuroblastoma: The French very rare tumors group (Fracture) contribution. Pediatr Blood Cancer 67 (4): e28154, 2020.
- Safi C, Spielman D, Otten M, et al.: Treatment Strategies and Outcomes of Pediatric Esthesioneuroblastoma: A Systematic Review. Front Oncol 10: 1247, 2020.
- Ozsahin M, Gruber G, Olszyk O, et al.: Outcome and prognostic factors in olfactory neuroblastoma: a rare cancer network study. Int J Radiat Oncol Biol Phys 78 (4): 992-7, 2010.
- Di Carlo D, Fichera G, Dumont B, et al.: Olfactory neuroblastoma in children and adolescents: The EXPeRT recommendations for diagnosis and management. EJC Paediatr Oncol 3: 100136, 2024. Also available online. Last accessed July 11, 2024.
- Soler ZM, Smith TL: Endoscopic versus open craniofacial resection of esthesioneuroblastoma: what is the evidence? Laryngoscope 122 (2): 244-5, 2012.
- Gallia GL, Reh DD, Lane AP, et al.: Endoscopic resection of esthesioneuroblastoma. J Clin Neurosci 19 (11): 1478-82, 2012.
- Unger F, Haselsberger K, Walch C, et al.: Combined endoscopic surgery and radiosurgery as treatment modality for olfactory neuroblastoma (esthesioneuroblastoma). Acta Neurochir (Wien) 147 (6): 595-601; discussion 601-2, 2005.
- Drescher NR, Indelicato DJ, Dagan R, et al.: Outcomes following proton therapy for pediatric esthesioneuroblastoma. Pediatr Blood Cancer 71 (2): e30793, 2024.
- Zanation AM, Ferlito A, Rinaldo A, et al.: When, how and why to treat the neck in patients with esthesioneuroblastoma: a review. Eur Arch Otorhinolaryngol 267 (11): 1667-71, 2010.
- Kumar M, Fallon RJ, Hill JS, et al.: Esthesioneuroblastoma in children. J Pediatr Hematol Oncol 24 (6): 482-7, 2002 Aug-Sep.
- Loy AH, Reibel JF, Read PW, et al.: Esthesioneuroblastoma: continued follow-up of a single institution's experience. Arch Otolaryngol Head Neck Surg 132 (2): 134-8, 2006.
- Porter AB, Bernold DM, Giannini C, et al.: Retrospective review of adjuvant chemotherapy for esthesioneuroblastoma. J Neurooncol 90 (2): 201-4, 2008.
- Benfari G, Fusconi M, Ciofalo A, et al.: Radiotherapy alone for local tumour control in esthesioneuroblastoma. Acta Otorhinolaryngol Ital 28 (6): 292-7, 2008.
- Kim DW, Jo YH, Kim JH, et al.: Neoadjuvant etoposide, ifosfamide, and cisplatin for the treatment of olfactory neuroblastoma. Cancer 101 (10): 2257-60, 2004.
- Kumar R: Esthesioneuroblastoma: Multimodal management and review of literature. World J Clin Cases 3 (9): 774-8, 2015.
- El Kababri M, Habrand JL, Valteau-Couanet D, et al.: Esthesioneuroblastoma in children and adolescent: experience on 11 cases with literature review. J Pediatr Hematol Oncol 36 (2): 91-5, 2014.
- Kiyota N, Tahara M, Fujii S, et al.: Nonplatinum-based chemotherapy with irinotecan plus docetaxel for advanced or metastatic olfactory neuroblastoma: a retrospective analysis of 12 cases. Cancer 112 (4): 885-91, 2008.
Opciones de tratamiento en evaluación clínica para el estesioneuroblastoma infantil
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Actualizaciones más recientes a este resumen (02 / 17 / 2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Este resumen fue objeto de revisión integral.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del estesioneuroblastoma infantil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del estesioneuroblastoma infantil son:
- Denise Adams, MD (Children's Hospital Boston)
- Karen J. Marcus, MD, FACR (Dana-Farber of Boston Children's Cancer Center and Blood Disorders Harvard Medical School)
- William H. Meyer, MD
- Paul A. Meyers, MD (Memorial Sloan-Kettering Cancer Center)
- Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
- Alberto S. Pappo, MD (St. Jude Children's Research Hospital)
- D. Williams Parsons, MD, PhD (Texas Children's Hospital)
- Arthur Kim Ritchey, MD (Children's Hospital of Pittsburgh of UPMC)
- Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
- Stephen J. Shochat, MD (St. Jude Children's Research Hospital)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre el tratamiento pediátrico. PDQ Tratamiento del estesioneuroblastoma infantil. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en: https://www.cancer.gov/espanol/tipos/cabeza-cuello/pro/infantil/tratamiento-estesioneuroblastoma-pdq. Fecha de acceso: <MM/DD/YYYY>.
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.
Última revisión: 2025-02-17

