Tumores benignos
Los tumores vasculares benignos son los siguientes:
- Hemangioma infantil.
- Hemangioma congénito.
- Tumores vasculares hepáticos.
- Hemangioma fusocelular.
- Hemangioma epitelioide.
- Granuloma piógeno (hemangioma capilar lobulillar).
- Angiofibroma.
- Angiofibroma nasofaríngeo juvenil.
El angiofibroma nasofaríngeo juvenil no se incluye en la clasificación de la Organización Mundial de la Salud (OMS) ni en la clasificación de la Internacional Society for the Study of Vascular Anomalies (ISSVA) de tumores vasculares. Se incluye aquí debido a que cada vez hay más evidencia que revela la diferenciación vascular y proliferación de estos tumores con respuesta a la remodelación vascular y los fármacos antiproliferativos.
Hemangioma infantil
Incidencia y epidemiología
Los hemangiomas infantiles (HI) son los tumores vasculares benignos más comunes de la primera infancia; se presentan en un 4 % a un 5 % de los lactantes. Se desconoce su verdadera incidencia.[1] No suelen estar presentes en el momento del nacimiento y se diagnostican con mayor frecuencia entre las 3 y 6 semanas de vida.[2,3,4,5] La lesión prolifera durante un promedio de 5 meses, se estabiliza y luego involuciona a lo largo de varios años.
Los hemangiomas infantiles son más comunes en las mujeres, los pacientes blancos que no son hispanos y los lactantes prematuros. Los hemangiomas múltiples son más comunes en los lactantes de gestaciones múltiples o de fertilización in vitro.[5,6,7] Los hemangiomas infantiles se relacionan con la edad avanzada de la madre, placenta previa, preeclampsia y otras anomalías placentarias.[5]
Cuadro clínico inicial
La mayoría de los hemangiomas infantiles no están presentes en el nacimiento, pero a menudo se observan lesiones precursoras como telangiectasia, discromía tenue o hipopigmentación de la piel. La lesión se puede confundir con un hematoma causado por trauma al nacer o con una malformación capilar (mancha en vino de Oporto) (consultar la Figura 1).[8,9]
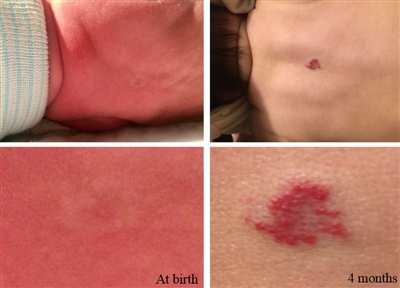
Los hemangiomas infantiles se pueden ubicar en la superficie de la dermis, en la profundidad del tejido subcutáneo, ser mixtos o estar en las vísceras. Las lesiones mixtas son comunes y con frecuencia se presentan en la cabeza y el cuello, pero a veces aparecen en cualquier lugar del cuerpo.
Los hemangiomas infantiles se caracterizan como sigue:
- Locales: la mayoría de las lesiones están localizadas y en un área bien definida sin evidencia de patrón geométrico.
- Segmentarios: la mayoría de los hemangiomas se presentan en la región de la cabeza y el cuello (Síndrome PHACE), pero es posible verlos en el área genitourinaria, los brazos, el tórax o las piernas (Síndrome LUMBAR/PÉLVICO/SACRO).
- Los hemangiomas difusos de la cara muestran patrones cutáneos definidos. En varios estudios se ha evaluado la distribución de estos hemangiomas y se encontraron los siguientes 4 patrones o segmentos característicos:
- El segmento 1 compromete las áreas lateral de la frente, temporal anterior del cuero cabelludo y frontal lateral del cuero cabelludo.
- Los segmentos 2 y 3 se encuentran en el área maxilar y mandibular.
- El segmento 4 cubre el área frontal medial del cuero cabelludo, la nariz y el surco nasolabial.
En 2 artículos, se informó sobre esta observación y se indica el compromiso de los derivados de la cresta neural en el hemangioma facial.[10,11] Los hemangiomas segmentarios por lo general se presentan en las mujeres y es más probable que estén relacionados con complicaciones y otros síndromes.[12,13]
Para obtener información sobre el síndrome PHACE o el síndrome LUMBAR/PÉLVICO/SACRO, consultar la sección Síndromes relacionados con el hemangioma infantil.
- Los hemangiomas difusos de la cara muestran patrones cutáneos definidos. En varios estudios se ha evaluado la distribución de estos hemangiomas y se encontraron los siguientes 4 patrones o segmentos característicos:
- Múltiples: más de 1 lesión, pero en el pasado se indicaba como más de 5 lesiones; esto se debe al aumento del riesgo de compromiso visceral (en su mayoría en el hígado).
El aspecto cutáneo de un hemangioma infantil por lo general es de color rojo o carmín, y es firme y caliente en la fase proliferativa. La lesión luego se aclara en el centro y se vuelve tibia y más suave; a continuación, se aplana y pierde el color. El proceso de involución puede demorar varios años y, después de involucionar, es poco común que vuelva a crecer. En 2 pacientes tratados con hormona del crecimiento, se observó nuevo crecimiento luego de la involución.[14] Después de realizar investigaciones adicionales, se encontraron receptores de hormona del crecimiento en las células del hemangioma infantil. Si bien es un resultado preliminar, este hallazgo quizás promueva la investigación sobre la etiología del crecimiento del hemangioma.
La ulceración es la complicación más frecuente de los hemangiomas infantiles, y se produce en el 10 % al 15 % de los pacientes. La ulceración suele aparecer durante la fase proliferativa y da lugar a hemorragias e infecciones secundarias.[15] La mayoría de las demás complicaciones en la fase proliferativa son consecuencia del efecto de la masa sobre las estructuras locales (por ejemplo, compromiso visual o auditivo, obstrucción de las vías respiratorias).[16]
Las secuelas permanentes que a veces se presentan después de la involución de los hemangiomas son la telangiectasia, la anetodermia, la piel redundante y un componente superficial persistente. Los hemangiomas con antecedentes de ulceración tienen más probabilidad de causar cicatrices y posibles complicaciones anatómicas locales.[15] En ausencia de ulceración, se describieron casos raros de disestesias en sitios de hemangiomas infantiles involucionados.[17][Nivel de evidencia C1] En un estudio de cohorte retrospectivo de 184 hemangiomas, la incidencia general de secuelas importantes fue del 54,9 %. Estas fueron más comunes en los hemangiomas mixtos, hemangiomas con borde elevado o irregular, y hemangiomas superficiales con aspecto de empedrado. Por otra parte, en este estudio se descubrió que la edad promedio de la involución del hemangioma fue de 3,5 años.[18]
Características biológicas e histopatológicas
La mayoría de los hemangiomas infantiles se presentan de modo esporádico. Sin embargo, en casos excepcionales obedecen a una anomalía del cromosoma 5 que se presenta con un modo de herencia autosómico dominante.[19] En un estudio en el que se evaluó el modo de herencia de los hemangiomas infantiles, se encontró que el 34 % de los pacientes tenía antecedentes familiares de hemangioma infantil, en su mayoría en un pariente de primer grado.[19,20]
Se desconoce el mecanismo exacto que produce la proliferación inicial de vasos sanguíneos seguida por la involución del componente vascular del hemangioma y el remplazo de tejido fibroadiposo. Se aislaron varios tipos de células de los hemangiomas: células madre o progenitoras (HemSC), células endoteliales (HemEC), pericitos (HemPericitos) y mastocitos.[21,22] Al parecer estas células intervienen en la formación del hemangioma infantil.
Las HemSC representan un porcentaje pequeño de las células del hemangioma proliferativo y tienen la capacidad de autorrenovación y diferenciación multilinaje. Estas células se diferencian y se convierten en células endoteliales, adipocitos o pericitos. Cuando las HemSC se implantaron en ratones con inmunodeficiencia, estas formaron lesiones similares a hemangiomas y luego presentaron regresión espontánea, parecido a lo que sucede con los hemangiomas infantiles.[23] Esto permite suponer que la proliferación del hemangioma infantil se produce durante la vasculogénesis (formación de vasos sanguíneos nuevos a partir de angioblastos), en lugar de la angiogénesis (formación de vasos sanguíneos nuevos a partir de vasos sanguíneos existentes).
Las HemEC son células hinchadas, con metabolismo activo y de apariencia semejante a las células endoteliales fetales en fase proliferativa. En la evaluación de las células endoteliales del hemangioma infantil se indica que estas son de naturaleza clonal.[23,24,25]
Los HemPericitos rodean la vasculatura y abundan en la fase proliferativa. Estas células expresan marcadores de pericitos y de células musculares lisas, como el antígeno neural/glial 2 (NG2), el receptor del factor de crecimiento derivado de plaquetas β (PDGFR β), calponina, actina α de músculo liso (SMA) y NOTCH3. Los HemPericitos son proangiogénicos ya que expresan aumento del factor de crecimiento endotelial vascular A (VEGF-A), disminución de la angiopoyetina-1 (ANGPT1), aumento de la proliferación, aumento de la formación de vasos sanguíneos in vivo y disminución de la capacidad de suprimir la proliferación.[26] En un estudio, se informó que los hemangiomas infantiles proliferativos contenían más concentraciones de RNA mensajero y proteínas para los receptores NOTCH1, 3 y 4 y sus ligandos, así como del coactivador MAML1 que la piel normal, los hemangiomas infantiles que involucionan y los hemangiomas infantiles tratados con propranolol.[27]
Hay una mayor cantidad de mastocitos durante la fase de involución temprana, pero también se encuentran en cantidades menores durante la fase proliferativa y al final de la involución. Se desconoce su función en los hemangiomas infantiles, aunque se ha observado que tienen cierta importancia en otros tumores cutáneos como el carcinoma de células basales, el carcinoma de células escamosas y el melanoma.[22]
Durante la proliferación, se expresan factores provasculogénicos, como el VEGF, el factor de crecimiento de fibroblastos (FGF), el CD34, el CD31, el CD133, el receptor hialurónico endotelial vascular linfático 1 (LYVE1) y el factor de crecimiento similar a la insulina 2 (IGF-2).[28,29,30,31] Durante la involución de los hemangiomas infantiles se observa aumento de la apoptosis.[31] Durante esta fase, también aumentan los mastocitos y las concentraciones de metaloproteinasa; asimismo, se produce un aumento regulado del interferón y disminuye el FGF básico (bFGF).[31,32,33] Durante la proliferación y la involución, las células endoteliales del hemangioma infantil exhiben un fenotipo particular en el que se observa una tinción positiva para antígenos de GLUT1 y aquellos relacionados con la placenta (receptores Fc γ II, merosina y antígeno Lewis Y). Estos marcadores están ausentes en los capilares normales y en otros tumores vasculares como el hemangioma congénito y las malformaciones vasculares. Las vellosidades coriónicas placentarias comparten estos mismos marcadores. Sin embargo, no se ha encontrado una relación entre estas y los hemangiomas.[28]
La hipoxia cumple una función importante en la patogenia de los hemangiomas. Los hemangiomas se relacionan con la hipoxia placentaria la cual aumenta con la prematuridad, los embarazos múltiples y las anomalías placentarias.[2,5] Se han observado diferentes dianas moleculares afectadas por hipoxia [34,35] en los hemangiomas proliferativos, como el VEGF-A, el GLUT1 y el IGF-2.[28,30,36] La hipótesis indica que un hemangioma proliferativo es un intento de normalizar el tejido hipóxico que se formó in utero.
Evaluación diagnóstica
Los hemangiomas se suelen diagnosticar por los antecedentes y el aspecto clínico. Pocas veces se necesita una biopsia y solo se realiza si el aspecto, los antecedentes y la presentación clínica inicial son atípicos. Por lo general, no se necesitan pruebas con imágenes, pero una ecografía diagnóstica es útil cuando hay una lesión más profunda sin un componente cutáneo que exhibe una lesión de flujo alto hipoecogénica bien circunscrita con una característica típica de onda Doppler.[37] Además, los lactantes con 5 o más hemangiomas cutáneos se deben someter a una ecografía de hígado para descartar la presencia de un hemangioma hepático.[38]
Hemangioma infantil de crecimiento mínimo o detenido
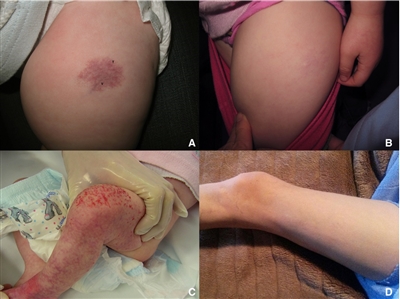
El hemangioma infantil de crecimiento mínimo o detenido (IH-MAG) es una variante del hemangioma que se puede confundir con una malformación capilar por sus características inusuales. Estos hemangiomas están completamente formados en el momento del nacimiento y se caracterizan por telangiectasia y vénulas con áreas de coloración claras y oscuras en la piel (consultar la Figura 2). Se resuelven de forma espontánea y son positivas para GLUT1 como característica patológica.[39] Se encuentran, en su mayoría, en la parte inferior del cuerpo pero a veces se presentan en la zona de la cabeza y el cuello. Si son segmentarios, es posible que se relacionen con el síndrome PHACE.[40] La hipertrofia del tejido blando vinculada a estos hemangiomas quizás persista durante la niñez.[41]
Hemangioma infantil de las vías respiratorias
Los hemangiomas infantiles de las vías respiratorias se suelen relacionar con hemangiomas segmentarios de distribución en barba, a veces incluye todas o algunas de las siguientes áreas: piel preauricular, mandíbula, labio inferior, mentón o parte anterior del cuello. Es importante que un otorrinolaringólogo evalúe de forma proactiva las lesiones con esta distribución antes de que aparezcan signos de estridor. La incidencia del hemangioma infantil de las vías respiratorias aumenta cuando hay compromiso de un área mayor de la barba.[42]
Es posible que en el hemangioma infantil de las vías respiratorias no se presenten lesiones en la piel. En un estudio retrospectivo de la Vascular Anomaly Database del Children's Hospital of Pittsburgh se analizaron 761 casos de hemangioma infantil. Entre estos, 13 pacientes (1,7 %) tenían hemangioma subglótico. De estos 13 pacientes, 4 (30 %) tenían distribuciones en barba, 2 (15 %) tenían hemangioma cutáneo y 7 (55 %) no tenían lesiones cutáneas.[43] Para obtener información sobre el tratamiento del hemangioma infantil de las vías respiratorias, consultar la sección Terapia con propranolol.
Compromiso oftálmico de los hemangiomas
Los hemangiomas periorbitarios a veces afectan la visión.[44] Por lo general, esto ocurre con los hemangiomas de la parte media del párpado superior; sin embargo, es posible que cualquier hemangioma alrededor de los ojos que sea lo bastante grande cause alteraciones en la córnea u obstrucciones en el eje visual. A veces los hemangiomas perioculares subcutáneos, se extienden a la órbita y producen exoftalmía o desplazamiento del globo ocular con manifestaciones cutáneas mínimas. Los problemas que producen estas lesiones incluyen astigmatismo debido a presión directa del hemangioma en crecimiento, ptosis, proptosis y estrabismo. Una de las causas más importantes de ceguera prevenible en niños es la ambliopía por privación de estímulos derivada de la obstrucción por un hemangioma. Se debe hacer una evaluación oftalmológica a todos los pacientes con hemangiomas periorbitarios o con cualquier posibilidad de compromiso visual.
En Francia y Canadá, 2 instituciones realizaron un análisis retrospectivo de pacientes con anomalías vasculares. Los investigadores revisaron los expedientes médicos de todos los pacientes con hemangioma infantil facial segmentario o periorbitario focal para los que se disponía de fotografías e imágenes por resonancia magnética (IRM) del encéfalo.[45][Nivel de evidencia C1] En el estudio participaron 122 pacientes (90 niñas, 32 niños; media de edad, 16,6 meses). De estos niños, 45 (36,9 %) presentaron un hemangioma infantil facial de más de 5 cm y 22 (18,0 %) tenían el síndrome PHACES o un posible síndrome PHACES. Se observaron anomalías cerebrovasculares estructurales en 14 de los 22 pacientes con el síndrome PHACES y en ninguno de los pacientes sin este síndrome. Se observaron anomalías encefálicas en 6 de 22 pacientes con el síndrome PHACES y en 1 paciente sin este síndrome (P < 0,001). Se observaron anomalías cardiovasculares en 6 pacientes y anomalías oculares en 8 pacientes. De estos 14 pacientes, 13 tenían el síndrome PHACES. Los autores concluyeron que la preocupación clínica sobre anomalías extracutáneas se justifica en todos los niños con hemangiomas infantiles periorbitarios focales o segmentarios, incluso en los que presentan hemangiomas pequeños.
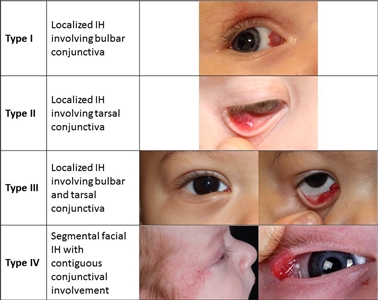
Los hemangiomas infantiles se pueden presentar en la conjuntiva (consultar la Figura 3). Estos hemangiomas a veces están relacionados con otras anomalías oftálmicas y se tratan con betabloqueantes orales o tópicos.[46]
Síndromes relacionados con el hemangioma infantil
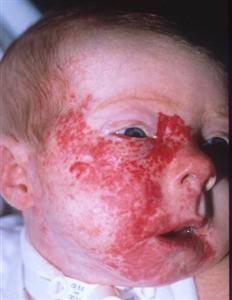
Síndrome PHACE
Malformaciones de la fosa posterior, hemangiomas, anomalías arteriales, cardíacas y oculares (Posterior fossa–brain malformations; Hemangiomas; Arterial, Cardiac, and Eye abnormalities): el síndrome PHACE (en inglés) es un grupo de enfermedades que se define por la presencia de hemangiomas infantiles segmentarios grandes, por lo general, en la cara o la cabeza, y a veces, en el cuello, el tórax y el brazo, que se relacionan con una o más malformaciones congénitas (consultar la Figura 4).[47] El síndrome PHACE es más frecuente en niñas y en neonatos de embarazos únicos nacidos a término y con peso normal.[13,48,49,50,51,52] El síndrome es frecuente en los pacientes con hemangiomas infantiles. En un estudio prospectivo de 108 lactantes con hemangiomas grandes en la cara, se observó que el 31 % de los pacientes sufría de síndrome PHACE.[53] Se han informado casos raros de síndrome PHACE en lactantes con hemangiomas de menos de 5 cm.[45][Nivel de evidencia C1]
En una reunión de un grupo de expertos, se actualizaron criterios de consenso para el síndrome PHACE definitivo y posible, de la siguiente manera:[47]
PHACE
- Anomalías de la fosa posterior. Las malformaciones de la fosa posterior, incluyen el complejo de Dandy-Walker, la hipoplasia cerebelosa, la atrofia y la disgenesia o agenesia del vermis. Los efectos de estas anomalías incluyen retrasos del desarrollo y disfunción hipofisaria.[54]
- Hemangiomas.[53,55,56,57,58]
- Un hemangioma segmentario grande en la cara o el cuero cabelludo con un área de superficie de 22 cm2 o mayor (5 cm × 4,5 cm).
- Un hemangioma segmentario grande en el cuello, tronco o extremidad superior proximal.
Los lactantes que cumplan con 2 criterios importantes para el síndrome PHACE (por ejemplo, el rafe supraumbilical y la coartación aórtica) pero que no tengan hemangiomas infantiles cutáneos deben someterse a una evaluación completa para evaluar la presencia de síndrome PHACE.
- Anomalías arteriales. Las anomalías cerebrovasculares incluyen anormalidades en la arteria carótida (incluso tortuosidad), y ausencia, dilatación y aneurisma o estrechamiento de los vasos encefálicos. Estas anomalías, en especial vinculadas a las arterias carótidas, producen obstrucción arterial progresiva e incluso accidente cerebrovascular. Las categorías de riesgo son las siguientes:[51,52,59,60,61]
- Riesgo bajo: esta categoría incluye pacientes con anomalías arteriales con frecuencia observadas en la población general a evaluar. También se incluyen hallazgos con poca repercusión clínica o ninguna en el desenlace del paciente, así sean infrecuentes en la población general. Algunos ejemplos de esto son las arterias embrionarias persistentes, el origen o curso arterial anómalo y las variaciones del polígono de Willis.
- Riesgo intermedio: se incluye a los pacientes con disgenesia sin estenosis; incluso aquellos con distensión o dilatación segmentaria de las arterias. También se incluye a los pacientes con estrechamiento u oclusión de las arterias proximales del polígono de Willis sin riesgo hemodinámico percibido. Es esencial hacer una evaluación de la permeabilidad del polígono de Willis.
- Riesgo alto: en esta categoría se incluye a los pacientes con 1 o más de las siguientes características:
- Estrechamiento considerable (>25 %) u oclusión de los vasos cerebrales principales ubicados dentro o encima del polígono de Willis y que produce una circulación aislada.
- Estenosis arterial múltiple o en tándem vinculada a una circulación sanguínea compleja que quizás produzca una disminución de la perfusión cerebral. Los pacientes con estenosis cerebrovascular en el entorno de una coartación aórtica tienen una probabilidad de riesgo más alto para presentar eventos isquémicos neurológicos permanentes y transitorios.
- Hallazgos en las imágenes del parénquima encefálico que indican isquemia crónica o asintomática, o enfermedad estenooclusiva progresiva. Estos hallazgos en las IRM del parénquima encefálico incluyen infarto existente, cambios isquémicos crónicos o de la zona marginal y presencia de dilatación de los vasos colaterales lentículoestriados o de los vasos colaterales de la piamadre.
- Anomalías cardíacas. Las anomalías del cayado aórtico que se observan en el síndrome PHACE suelen ser complejas y afectan la sección transversal y descendente del cayado. Esta obstrucción es casi siempre de segmentos largos. Por lo general, la obstrucción se caracteriza por la presencia de áreas estrechas del cayado con segmentos adyacentes de dilatación aneurismática marcada.
- Anomalías oculares. Las anomalías oculares incluyen la microftalmia, las anomalías vasculares retinianas y los vasos retinianos fetales persistentes, la exoftalmía, el coloboma y la atrofia del nervio óptico. Estas anomalías son poco comunes y se presentan entre el 7 % y el 10 % de los pacientes.[62]
En una revisión retrospectiva, se identificaron hamartomas mesenquimatosos rabdomiomatosos en la línea media y hamartomas en el mentón en una cantidad pequeña de niños con el síndrome PHACE o el síndrome LUMBAR.[63] En la actualidad, estos no se incluyen como criterios menores.
Para diagnosticar el síndrome PHACE se necesita un examen clínico, una evaluación cardíaca con ecocardiograma, una evaluación oftalmológica, e imágenes por resonancia magnética (IRM) o angiografía por resonancia magnética (ARM) de la cabeza y el cuello. Todos los pacientes con hallazgos de riesgo intermedio y de riesgo alto en el sistema nervioso central (SNC) necesitan el seguimiento de un neurólogo o neurocirujano. Una coartación aórtica requiere de consulta cardiológica inmediata y quizás se justifique una IRM o ARM. En un informe de 2 pacientes con hemangiomas infantiles retrorbitales y arteriopatía, se indicó una posible presentación inicial nueva del síndrome PHACE.[58] Para los pacientes con proptosis, desviación del globo ocular y estrabismo se recomienda una IRM o ARM. Es posible que se necesiten pruebas adicionales para el síndrome PHACE según los hallazgos en el sistema nervioso central (SNC).
Los problemas a corto y largo plazo relacionados con el síndrome PHACE son los siguientes:[64,65,66]; [67][Nivel de evidencia C1]
- Cefalea.
- En ocasiones se presenta a una edad temprana.
- Puede ser intensa.
- Las cefaleas de nueva aparición deben evaluarse para descartar vasculopatía o isquemia cerebral.
- Se recomienda la derivación a un neurólogo.
- Los medicamentos vasoconstrictores están contraindicados.
- Hipoacusia y retraso en el habla y el lenguaje.
- Los retrasos en el habla y el lenguaje pueden ser consecuencia de deficiencias auditivas, hospitalizaciones prolongadas u otras anomalías del desarrollo neurológico.
- La hipoacusia neurosensitiva es el tipo más común y suele ser ipsilateral al hemangioma infantil, lo que quizás afecte el VIII par craneal ipsilateral.
- La detección temprana es fundamental.
- Todos los pacientes con síndrome PHACE deben someterse a exámenes de detección auditivos cuando nacen y al menos durante una visita de seguimiento si el examen de detección inicial es normal.
- Disfagia, trastornos de la alimentación, trastornos del habla y retraso del lenguaje.
- Más acentuado en pacientes con malformaciones de la fosa posterior, hemangiomas de labio u orofaringe o hemangiomas de las vías respiratorias, hipoacusia, y aquellos con antecedentes de cirugía cardíaca.
- Los pacientes deben someterse a una evaluación inicial del habla y el lenguaje antes de los 24 meses de edad.
- Los pacientes con dificultades de alimentación deben derivarse a un foniatra pediátrico para una evaluación a cualquier edad.
- La disfagia a veces es secundaria al sitio de la enfermedad (labio, cavidad oral y faringe) o a la motricidad oral.
- Anomalías endocrinas.
- Las anomalías que se notifican con más frecuencia son la disfunción tiroidea y el hipopituitarismo que produce deficiencia de la hormona de crecimiento.
- También se han descrito otras manifestaciones de hipopituitarismo, como hipogonadismo hipogonadotrópico e insuficiencia suprarrenal.
- Los pacientes deben someterse a un examen de detección neonatal y se tiene que repetir el estudio si se presentan síntomas.
- Deficiencia de la hormona de crecimiento: la mayoría de los casos notificados se vinculan a hipopituitarismo con silla turca vacía o parcialmente vacía, según se observa en la IRM, pero también se producen sin evidencia de malformaciones en el sistema nervioso central.
- La hipoglucemia neonatal puede ser un signo de hipopituitarismo y es motivo de evaluación endocrinológica adicional.
- Otras consecuencias de la disfunción de la hipófisis son el hipogonadismo hipogonadotrópico, que se manifiesta mediante un retraso en el inicio de la pubertad e insuficiencia suprarrenal de inicio tardío. Estos hallazgos resaltan la importancia de centrar la evaluación en la altura, el peso y los hitos del desarrollo durante la atención de los niños con síndrome PHACE.
- Anomalías dentales (hipoplasia adamantina).
- En un estudio de 18 niños con síndrome PHACE, o con posibilidades de presentar este síndrome, se observó que el 28 % de los pacientes presentaba hipoplasia adamantina. Todos los niños afectados presentaban hemangiomas intraorales. Cinco de los 11 pacientes (45 %) con hemangiomas intraorales presentaban defectos adamantinos. Los niños con hipoplasia adamantina tienen un mayor riesgo de caries.
- Se debe evaluar a los pacientes para descartar la presencia de hemangiomas intraorales. Si se detectan, los pacientes se deben remitir a un dentista pediátrico antes de cumplir 1 año para la detección y el tratamiento tempranos.
- Desenlaces a largo plazo y calidad de vida.
- Un grupo internacional de expertos publicó un informe de un estudio multicéntrico en el que se usó un método transversal con entrevistas y revisiones de las historias clínicas para analizar los desenlaces a largo plazo y la calidad de vida de los pacientes con síndrome PHACE de 10 años o más.[68] El diagnóstico definitivo de síndrome PHACE se hizo según guías publicadas antes.[47] Esta ha sido la cohorte más grande de adolescentes y adultos con síndrome PHACE. Se contactó a 153 pacientes y de ellos 104 participaron en el estudio (68 %). La mediana de edad fue de 14 años (intervalo, 10–77 años). En este estudio se encontró que el síndrome PHACE se asocia con complicaciones mórbidas a largo plazo de leves a graves, como hemangioma infantil residual (94,1 %), cefaleas o migrañas (72,1 %), diferencias en el aprendizaje (45,1 %), y arteriopatía progresiva (29,4 %). En el Cuadro se describen otros resultados del estudio.
La mayoría de los pacientes con hemangioma residual estaban satisfechos o muy satisfechos con su apariencia (89,5 %). Fue menos probable que aquellos con antecedentes de cirugía o ulceración notificaran un efecto mínimo en su autoconfianza. De 68 pacientes con arteriopatía que contaban con imágenes de seguimiento, 6 (8,8 %) presentaron vasculopatía moyamoya o esteno-oclusión progresiva que llevó a una circulación aislada a nivel del polígono de Willis o por encima de este. A pesar de esos hallazgos, la proporción de pacientes con ictus isquémico fue baja (2 de 104; 1,9 %). Las puntuaciones de salud general del Patient-Reported Outcomes Measurement Information System (PROMIS) fueron más bajas que los valores normales de la población por lo menos en 1 desviación estándar. Debido a la prevalencia general del síndrome PHACE, no fue posible obtener una potencia estadística suficiente para evaluar de manera exacta todos los desenlaces. Los autores del estudio concluyeron que para los pacientes con síndrome PHACE el seguimiento primario y especializado es importante hasta la edad adulta. Se necesitan nuevos estudios para identificar las orientaciones precisas para hacer el seguimiento a largo plazo.[68]
Cuadro 3. Hallazgos adicionales de la cohorte de personas con síndrome PHACEa Síntomas Prevalencia Síntomas Prevalencia TDAH = trastorno por déficit de atención e hiperactividad. a Adaptación de: Mitchell Braun, Ilona J. Frieden, Dawn H. Siegel, Elizabeth George, Christopher P. Hess, Christine K. Fox, Sarah L. Chamlin, Beth A. Drolet, Denise Metry, Elena Pope, Julie Powell, Kristen Holland, Caden Ulschmid, Marilyn G. Liang, Kelly K. Barry, Tina Ho, Chantal Cotter, Eulalia Baselga, David Bosquez, Surabhi Neerendranath Jain, Jordan K. Bui, Irene Lara-Corrales, Tracy Funk, Alison Small, Wenelia Baghoomian, Albert C. Yan, James R. Treat, Griffin Stockton Hogrogian, Charles Huang, Anita Haggstrom, Mary List, Catherine C. McCuaig, Victoria Barrio, Anthony J. Mancini, Leslie P. Lawley, Kerrie Grunnet-Satcher, Kimberly A. Horii, Brandon Newell, Amy Nopper, Maria C. Garzon, Margaret E. Scollan, Erin F. Mathes, Multicenter Study of Long-Term Outcomes and Quality of Life in PHACE Syndrome after Age 10, The Journal of Pediatrics, Volumen 267, 2024, 113907, ISSN 0022-3476,https://doi.org/10.1016/j.jpeds.2024.113907. Este es un artículo de acceso libre distribuido bajo los términos de la licenciaCreative Commons CC-BY, que permite el libre uso, distrubución y reproducción por cualquier medio siempre y cuando se cite de manera apropiada el trabajo original. b Notificación de por lo menos una convulsión. c Incluye el síndrome deTourette, los temblores intencionales (intention tremorintention tremor) y el trastorno de movimiento de origen psicógeno (movement disordermovement disorder). Crecimiento tardío de un hemangioma infantil 13/104 (12,5 %) Dificultades visuales 56/104 (53,8 %) Intensificación del color 11/13 (84,6 %) Ceguera legal unilateral 5/104 (4,8 %) Crecimiento profundo 2/13 (15,4 %) Cirugías oculares 26/104 (25 %) Aumento del volumen 6/13 (46,2 %) Hipoacusia 18/104 (17,3 %) Otros síntomas neurológicos Hipoacusia conductiva 3/18 (16,7 %) Convulsionesb 15/104 (14,4 %) Hipoacusia neurosensorial 3/18 (16,7 %) Dificultades del habla 36/104 (34,6 %) Hipoacusia mixta 6/18 (33,3 %) Tratamiento de fonoaudiología 30/104 (28,8 %) Hipoacusia no clasificada 3/18 (16,7 %) Problemas de equilibrio 28/104 (26,9 %) Uso de audífonos 12/104 (11,5 %) Dificultad para deglutir 11/104 (10,6 %) Problemas dentales Tics nerviososc 6/104 (5,8 %) Problemas de raíz dental 16/104 (15,4 %) Diagnósticos relacionados con el aprendizaje Alteraciones del esmalte 31/104 (29,8 %) TDAH 19/104 (18,3 %) Dislexia 10/104 (9,6 %)
- Un grupo internacional de expertos publicó un informe de un estudio multicéntrico en el que se usó un método transversal con entrevistas y revisiones de las historias clínicas para analizar los desenlaces a largo plazo y la calidad de vida de los pacientes con síndrome PHACE de 10 años o más.[68] El diagnóstico definitivo de síndrome PHACE se hizo según guías publicadas antes.[47] Esta ha sido la cohorte más grande de adolescentes y adultos con síndrome PHACE. Se contactó a 153 pacientes y de ellos 104 participaron en el estudio (68 %). La mediana de edad fue de 14 años (intervalo, 10–77 años). En este estudio se encontró que el síndrome PHACE se asocia con complicaciones mórbidas a largo plazo de leves a graves, como hemangioma infantil residual (94,1 %), cefaleas o migrañas (72,1 %), diferencias en el aprendizaje (45,1 %), y arteriopatía progresiva (29,4 %). En el Cuadro se describen otros resultados del estudio.
Síndrome LUMBAR/PÉLVICO/SACRO
Los hemangiomas localizados sobre la columna lumbar o sacra quizá se relacionen con problemas genitourinarios, anomalías anorrectales o problemas neurológicos como la médula anclada.[69,70,71,72] Para describir el síndrome de hemangioma segmentario infantil en las áreas lumbar, pélvica y sacra, se han utilizado los siguientes criterios. Este síndrome se ha descrito en la bibliografía con varios acrónimos.
LUMBAR
- Hemangiomas de la parte inferior del cuerpo y otros defectos cutáneos (L ower-body hemangiomas and other cutaneous defects).
- Anomalías o ulceraciones urogenitales (U rogenital anomalies or ulceration).
- Mielopatía (M yelopathy).
- Deformidades óseas (B ony deformities).
- Malformaciones o anomalías arteriales anorrectales (A norectal malformations or arterial anomalies).
- Anomalías renales (R enal anomalies).
PELVIS
- Hemangiomas perineales (P erineal hemangiomas).
- Malformaciones de genitales externos (E xternal genital malformations).
- Lipomielomeningocele (L ipomyelomeningocele).
- Anomalías vesicorrenales (V esicorenal abnormalities).
- Ano imperforado (I mperforate anus).
- Papiloma cutáneo (S kin tag).
SACRO
- Disrrafia espinal (S pinal dysraphism).
- Malformaciones anogenitales (A nogenital).
- Anomalías cutáneas (C utaneous).
- Anomalías renales y urológicas (R enal and urologic anomalies) relacionadas con un angioma (A ssociated with an angioma) localización lumbosacral (L umbosacral localization).
Las lesiones segmentarias sobre el pliegue interglúteo y la columna lumbar se deben evaluar con ecografía o IRM, según la edad del paciente. En varios estudios, las evaluaciones con ecografías no identificaron algunas anomalías vertebrales que después se detectaron en la evaluación con IRM.[73,74]
Hemangiomas múltiples
Los lactantes con más de 5 hemangiomas se deben evaluar para descartar la presencia de hemangiomas viscerales. El sitio de compromiso más común es el hígado, en donde es posible observar lesiones múltiples o difusas.[75,76,77] A menudo, estas lesiones son asintomáticas pero, en una minoría de casos, se presentan síntomas de insuficiencia cardíaca secundaria a derivaciones de los vasos grandes, síndrome compartimental o hipotiroidismo profundo por la expresión de la yodotironina desyodinasa en las células de hemangioma.[78] Se pueden presentar hemangiomas hepáticos múltiples o difusos sin lesiones cutáneas. Otras posibles complicaciones poco frecuentes de los hemangiomas viscerales dependen del compromiso de órganos específicos y derivan de los efectos de la masa tumoral. Estas complicaciones son la hemorragia gastrointestinal, la ictericia obstructiva y las secuelas en el SNC. Para obtener más información, consultar la sección Tumores vasculares hepáticos.
Tratamiento del hemangioma infantil
La decisión de tratar a los pacientes con hemangiomas se fundamenta en varios factores, como los siguientes:[79]
- Tamaño de las lesiones.
- Tipo de hemangioma.
- Localización del hemangioma.
- Presencia o riesgo de complicaciones, como ulceración, probabilidad de dejar cicatriz o causar desfiguración, edad del paciente y estadio del crecimiento del hemangioma.
Esta decisión se hace de manera individual para cada paciente y es importante evaluar con detenimiento los riesgos y beneficios del tratamiento.
La American Academy of Pediatrics publicó guías de práctica clínica sobre este tema. Se observó que una intervención terapéutica temprana era importante para prevenir las complicaciones médicas y la desfiguración permanente producidas por los hemangiomas infantiles complejos. Se indicó que el mejor momento para llevar a cabo las intervenciones es en los primeros 1 a 3 meses de vida. Se usaron fotografías para clasificar los hemangiomas de bajo riesgo versus alto riesgo,[80] y se usó un sistema de puntuación para motivar a que los médicos de atención primaria hagan la derivación temprana a especialistas en hemangiomas.[81] Las guías indican que los especialistas en hemangiomas son los médicos expertos en el tratamiento y cuidado de hemangiomas y son quienes tienen el conocimiento sobre la estadificación de los riesgos y las opciones de tratamiento. Este grupo de proveedores está formado por expertos en los campos de dermatología, hematología y oncología, pediatría, cirugía plástica, cirugía general, otorrinolaringología y oftalmología.[82]
Las opciones de tratamiento para el hemangioma infantil son las siguientes:
- Terapia con propranolol.
- Terapia con betabloqueantes selectivos y de otro tipo.
- Terapia con corticoesteroides.
- Terapia láser.
- Por lo general, se utiliza para los hemangiomas ulcerados y las lesiones residuales como una telangiectasia después del período de proliferación.[83] La terapia con láser de colorante pulsado ayuda a controlar el dolor de los hemangiomas infantiles ulcerados. El uso de esta terapia de láser como tratamiento inicial para los hemangiomas infantiles es objeto de controversia.
- En un estudio piloto ruso, se utilizó un equipo láser multilínea con emisores Nd:YAP Q-Sw/KTP combinados con 2 longitudes de onda de 1079 nm y 540 nm para el tratamiento de pacientes con hemangiomas infantiles.[84] Se trataron con láser 109 pacientes que tenían 119 hemangiomas. La evaluación de las muestras después del tratamiento reveló la restauración del color normal, alivio de la piel y ausencia de cicatrices.
- En un estudio retrospectivo realizado en China se incluyó a 180 pacientes con hemangiomas superficiales que recibieron terapia con láser de colorante pulsado de 595 nm. En el estudio se notificó que los niños más pequeños (edad <2 meses) recibieron menos tratamientos, tuvieron cursos más cortos de la enfermedad y experimentaron efectos mejores con menos eventos adversos, en comparación con los niños mayores.[85]
- Escisión quirúrgica. Con el advenimiento de nuevos tratamientos médicos, la cirugía se reserva para lesiones ulceradas, lesiones residuales, lesiones perioculares grandes que interfieren con la visión y lesiones en la cara con un efecto cosmético que no responden a la terapia médica.[86]
- Terapia con betabloqueantes tópicos.
- Terapia combinada para los hemangiomas complicados.
Terapia con propranolol
El propranolol, un betabloqueante no selectivo, es una terapia de primera línea para los hemangiomas infantiles. En estudios tempranos se indica que el propranolol quizás actúa al inducir vasoconstricción o disminución de la expresión de VEGF y bFGF, lo que conduce a la apoptosis.[87,88] En estudios posteriores se indica que la actividad del propranolol en los hemangiomas infantiles no es secundaria a los betabloqueantes resultantes de la acción del enantiómero S(-) del propranolol, sino que resulta de la capacidad del enantiómero R(+) del propranolol de inhibir SOX18, un factor de transcripción que actúa como regulador principal de la vasculogénesis.[89,90,91] El enantiómero R(+) interfiere con la activación transcripcional de SOX18, interrumpe la dinámica de unión entre la cromatina y SOX18 e inhibe la formación de dímeros de SOX18. Estos efectos bioquímicos resultan en la inhibición de la diferenciación de las células madre del hemangioma en células endoteliales y en la inhibición de la vasculogénesis.[91]
El efecto del propranolol se observó por primera vez en dos lactantes tratados por problemas cardíacos en Europa. Se observó cambio de color, ablandamiento y disminución del tamaño del hemangioma. Desde entonces, se notificaron los resultados de un ensayo aleatorizado controlado.[92] En 2014, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó Hemangeol, la formulación pediátrica de clorhidrato de propranolol, para el tratamiento de los hemangiomas infantiles proliferativos. El uso del propranolol genérico sigue siendo frecuente.
Hay muchos otros informes publicados sobre la eficacia e inocuidad del propranolol.[93,94,95,96,97] La ausencia de respuesta al tratamiento es infrecuente. La terapia con propranolol por lo general se usa durante la fase de proliferación, pero también es eficaz para pacientes mayores de 12 meses con hemangiomas infantiles.[98]; [99][Nivel de evidencia C3]
Evidencia (terapia con propranolol):
- En un ensayo aleatorizado grande patrocinado por la industria, 456 lactantes de 5 semanas a 5 meses de edad con hemangiomas infantiles proliferativos de por lo menos 1,5 cm recibieron placebo o propranolol (1 mg/kg por día o 3 mg/kg por día) durante 3 o 6 meses. Después de un análisis provisional de los primeros 188 pacientes que completaron 24 semanas del tratamiento del ensayo, se seleccionó el régimen de 3 mg/kg por día durante 6 meses para el análisis final de eficacia.[92][Nivel de evidencia B3]
- De los pacientes que recibieron el régimen seleccionado, el 88 % mostraron mejoría en la semana 5, en comparación con el 5 % en los pacientes que recibieron el placebo.
- Los efectos adversos fueron poco frecuentes.
- En un estudio retrospectivo de 635 lactantes con hemangiomas infantiles tratados con propranolol (2 mg/kg por día), se obtuvieron los siguientes resultados:[97][Nivel de evidencia C3]
- La tasa de respuesta general fue del 91 %, y se observó regresión en la mayoría de los pacientes.
- El 2 % de los pacientes presentó efectos secundarios, ninguno de estos fue grave.
- En un metanálisis en el que se evaluaron 5130 pacientes de 61 estudios, se concluyó que el propranolol fue más eficaz e inocuo que otros tratamientos para el hemangioma infantil.[100]
- Las lesiones por hemangioma infantil de las vías respiratorias son infrecuentes. En un metanálisis de 61 pacientes, se informó los siguientes resultados:[101]
- Hubo una tendencia a la disminución del fracaso del tratamiento con estrategias de aumento de dosis, lo que es compatible con el uso de dosis más altas de propranolol para estos pacientes (3 mg/kg/día).
- En el análisis también se indicó que el uso simultáneo de corticoesteroides y propranolol quizás haya reducido la eficacia en los pacientes con hemangiomas infantiles segmentarios de las vías respiratorias; sin embargo, los tratamientos anteriores con corticoesteroides no tuvieron un efecto perjudicial.
- Se necesitan más estudios prospectivos para validar estos hallazgos.
- Los hemangiomas difusos (segmentarios) de vías respiratorias son muy raros y su comportamiento clínico es diferente al de las lesiones de vías respiratorias aisladas.
La administración intralesional de propranolol se usa para las lesiones periorbitarias en una capacidad limitada y no se observan ventajas en comparación con la administración oral.[102][Nivel de evidencia B3]
Se informó sobre varias recomendaciones del comité de consenso de expertos, incluso sobre las recomendaciones de la FDA y la European Medicines Agency después de un ensayo controlado aleatorizado del propranolol oral en pacientes con hemangioma infantil que condujo a la aprobación de la FDA.[103,104,105]
Las consideraciones para el uso del propranolol son las siguientes:[103,105,106]
- Inicio del tratamiento: las guías de los comités de consenso indican que el tratamiento se debe llevar a cabo en consulta con un especialista en anomalías vasculares pediátricas con pericia en el diagnóstico y el tratamiento de los tumores vasculares infantiles, y en el uso de propranolol en los niños. Recomiendan que se considere la hospitalización cuando se inicia la administración de propranolol oral bajo las siguientes circunstancias:[103]
- Lactantes de 4 semanas de edad o menos (corregida por la edad gestacional).
- Lactante de cualquier edad con apoyo social insuficiente.
- Lactante de cualquier edad con afecciones comórbidas que tengan efectos en el sistema cardiovascular o respiratorio, incluso hemangiomas de las vías respiratorias sintomáticos.
- Lactante de cualquier edad con afecciones que afecten la regulación de la glucemia.
La evaluación pretratamiento (paciente hospitalizado o ambulatorio) incluye los siguientes aspectos:
- Antecedentes, con especial atención a las anomalías cardiovasculares y respiratorias (por ejemplo, problemas de alimentación, disnea, taquipnea, diaforesis, sibilancias, soplo cardíaco), y antecedentes familiares de bloqueo cardíaco o arritmia.
- Examen físico que incluya una evaluación cardíaca y pulmonar, así como la medición de la frecuencia cardíaca.
- Los pacientes de riesgo estándar no necesitan un ecocardiograma o electrocardiograma. En 2 estudios, no se encontraron contraindicaciones para la terapia con betabloqueantes en el 6,5 % al 25 % de los pacientes con anomalías en un electrocardiograma.[106,107] Se debe considerar un electrocardiograma en niños con frecuencia cardíaca más baja de la normal para la edad y antecedentes de arritmia o arritmia detectada durante el examen.
- Antecedentes familiares de enfermedad cardíaca congénita o antecedentes maternos de enfermedades del tejido conjuntivo.
- Dosificación: según los paneles de consenso, por lo general, la dosificación que se usa es de 1 a 3 mg/kg por día dividida en 2 o 3 dosis. La dosis inicial varía según los factores de riesgo y la localización donde se administra por primera vez. Los pacientes ambulatorios y hospitalizados inician con una dosis de 0,5 mg/kg por día a 1 mg/kg por día la cual se va aumentando.[104,105,106] En lactantes menores de 5 semanas y pacientes con síndrome PHACE, se recomienda empezar con la dosificación de 3 veces por día.[47,103]
- Vigilancia: la vigilancia varía según la institución donde se administre. Sin embargo, el propranolol oral alcanza su valor máximo después de 1 a 3 horas de la administración; en la mayoría de los centros se mide la frecuencia cardíaca y la presión sanguínea arterial 1 y 2 horas después de cada dosis y, luego, cuando se aumenta la dosis por lo menos 0,5 mg/kg por día. La educación de los padres y el paciente incluye cuándo interrumpir el medicamento, los síntomas de hipoglucemia, la alimentación necesaria durante la noche y cuándo se deben comunicar con el médico por problemas, como una enfermedad que quizás interfiera con la ingesta oral o que produzca deshidratación o dificultades respiratorias.
En un estudio multicéntrico retrospectivo grande, se analizó la inocuidad de la administración ambulatoria de propranolol y se evaluó la necesidad de la vigilancia. En este estudio, se evaluó a 783 pacientes con 1148 consultas. No se observó bradicardia o hipotensión sintomáticas. La evaluación de la presión sanguínea no fue confiable. Los resultados indicaron la posibilidad de que la evaluación ambulatoria no sea necesaria en los pacientes de riesgo estándar con hemangiomas infantiles.[108]
- Contraindicaciones: la terapia con propranolol está contraindicada en lactantes y niños que tienen las siguientes afecciones:[103,104,105]
- Bradicardia sinusal.
- Hipotensión.
- Bloqueo auriculoventricular mayor de primer grado.
- Insuficiencia cardíaca.
- Asma.
- Hipersensibilidad.
- Síndrome PHACE. Es posible que el síndrome PHACE con arteriopatía en el SNC o con coartación de la aorta sea una contraindicación relativa. En un estudio multinstitucional retrospectivo que investigó la inocuidad del tratamiento con propranolol en pacientes con el síndrome PHACE, se identificó a 76 lactantes, incluso 12 pacientes con riesgo alto de presentar un accidente cerebrovascular.[109] La incidencia de efectos adversos en estos pacientes fue similar a la incidencia en 726 lactantes que recibieron tratamiento con propranolol oral para un hemangioma pero no cumplían los criterios para el síndrome PHACE. La decisión de tratar se debe tomar en consulta con neurología o neurocirugía y cardiología.
- Efectos adversos: los efectos adversos del propranolol se indican a continuación.[110]
- Hipoglucemia.
En un estudio realizado en Japón se hizo un seguimiento de la hipoglucemia en lactantes con hemangiomas infantiles que iniciaron tratamiento con propranolol.[111] Tras el tratamiento con propranolol, las incidencias de hipoglucemia grave y convulsiones hipoglucémicas fueron de alrededor del 0,54 % y el 0,35 %, respectivamente. La incidencia de convulsiones hipoglucémicas era mayor en Japón que en los países occidentales. La hipoglucemia grave fue frecuente en lactantes menores de 1 año cuando se usó propranolol durante 6 meses o más. La hipoglucemia grave se presentó con frecuencia entre las 5:00 y las 9:00 de la mañana, y se relacionó a menudo con períodos prolongados de ayuno, mala alimentación o condiciones físicas precarias.
- Hipotensión.
- Bradicardia.
- Trastornos del sueño.
- Diarrea o estreñimiento.
- Extremidades frías.
Estas complicaciones se notificaron en varios estudios y las complicaciones graves fueron infrecuentes.[110,112] El riesgo de estas complicaciones aumenta en pacientes con comorbilidades y enfermedades simultáneas, como diarrea, vómitos e infecciones respiratorias. Se debe considerar la necesidad de vigilancia estricta y posibles períodos de interrupción del medicamento durante períodos de enfermedad.
En una revisión retrospectiva de 1260 niños con hemangiomas infantiles tratados con propranolol, se identificaron 26 pacientes (2,1 %) con efectos secundarios que necesitaron la interrupción del propranolol.[113] La razón más común para la interrupción del propranolol fue el trastorno del sueño grave, que representó el 65,4 % de los casos. En total, 23 pacientes recibieron atenolol y 3 pacientes recibieron prednisolona como tratamiento de segunda línea. En los análisis multivariables solo la edad joven (intervalo de confianza [IC] 95 %, 1,201–2,793; P = 0,009) y el peso corporal bajo (IC 95 %, 1,036–1,972; P = 0,014) se relacionaron con la intolerancia a los efectos secundarios.
- Hipoglucemia.
- Duración del tratamiento: no hay pautas consensuadas para la duración del tratamiento con propranolol. En un estudio multinstitucional prospectivo que analizó la eficacia e inocuidad del propranolol en pacientes de riesgo alto, la administración de propranolol por 6 meses como mínimo, en niños de hasta 12 meses, aumentó el éxito del tratamiento; la dosis de este medicamento fue de 3 mg/kg por día. Los resultados del tratamiento se mantuvieron hasta 3 meses después de interrumpir la terapia. La eficacia y la inocuidad del propranolol en este estudio fueron similares a las que se notificaron en otros estudios.[114]
- Reactivación del hemangioma tras la terapia con propranolol: la reactivación es el crecimiento de hemangiomas infantiles después del cese de la administración de propranolol. En una revisión multinstitucional retrospectiva de 997 pacientes de hemangioma infantil, se observó una tasa de reactivación del 25,3 % en 912 pacientes para los que se contó con datos suficientes. En un análisis univariante, los factores relacionados con la reactivación incluyeron la interrupción del tratamiento antes de los 9 meses de edad, el sexo femenino, la localización en la cabeza o el cuello, el patrón segmentario y el compromiso de piel profunda o mixta. En un análisis multivariante, solo se observó una relación significativa entre los hemangiomas infantiles profundos y el sexo femenino.[115] En una revisión retrospectiva de un solo centro, se examinó a 198 pacientes con hemangioma infantil que recibieron propranolol oral. En el estudio se notificó que 35 pacientes (18 %) presentaron reactivación 1 a 3 meses después de la interrupción del tratamiento con propranolol. De los 35 pacientes, 23 volvieron a recibir propranolol durante un máximo de 3 meses. Todos los pacientes obtuvieron buenas respuestas.[116][Nivel de evidencia C3]
- Crecimiento tardío de los hemangiomas infantiles: es posible que el crecimiento de un hemangioma se presente en pacientes mayores de 3 años y se ha informado de su crecimiento hasta los 8,5 años. Los factores de riesgo relacionados incluyen la morfología segmentaria, los hemangiomas grandes, el síndrome PHACE y las lesiones cutáneas y subcutáneas profundas en la cabeza y el cuello.[117,118]
Terapia con betabloqueantes selectivos y de otro tipo
Debido a la naturaleza no selectiva y lipofílica del propranolol y su capacidad de pasar la barrera hematoencefálica, se están usando otros betabloqueantes para tratar los hemangiomas infantiles.
Evidencia (terapia con betabloqueantes):
- En 2 pequeños estudios comparativos, no hubo diferencia en la eficacia entre el propranolol y el atenolol.[119,120]
- Para respaldar un estudio retrospectivo anterior, en un estudio prospectivo con enmascaramiento doble se comparó el nadolol con el propranolol en 71 lactantes (de 1 a 6 meses de edad).[121][Nivel de evidencia A3]
- En el estudio se demostró la ausencia de inferioridad con respecto a la eficacia y el tratamiento.
- En 1 estudio prospectivo de 76 lactantes tratados con atenolol, se observó eficacia e inocuidad similares a las del propranolol.[122][Nivel de evidencia C3]
En un informe publicado, se relacionó el uso de nadolol con la muerte de un lactante (edad, 17 semanas) luego de 10 días sin producción de materia fecal.[123] Hay poca información sobre la farmacocinética y la inocuidad del nadolol en lactantes. Este fármaco tiene un índice terapéutico estrecho y se elimina en las heces sin sufrir alteraciones. Si se administra nadolol a un lactante, es fundamental la vigilancia para controlar la producción regular de materia fecal.
Se necesitan más estudios para evaluar las diferencias de toxicidad entre estos fármacos y el propranolol.
Hay algunos indicios de que los betabloqueantes más selectivos producen menos efectos secundarios.[124] En 1 estudio, se indicó la posibilidad de que el enantiómero (R+) del propranolol, que se transfiere en la síntesis del fármaco, en lugar del enantiómero antiadrenérgico β L(-) (el fármaco disponible para la venta es una mezcla racémica), produzca un efecto terapéutico contra el hemangioma infantil.[89,90]
Terapia con corticoesteroides
Antes del propranolol, los corticoesteroides eran el tratamiento de primera línea para el hemangioma infantil. Se usaron por primera vez al final de la década de 1950, pero nunca recibieron la aprobación de la FDA de los Estados Unidos para esta indicación. La terapia con corticoesteroides se tornó menos popular debido a los efectos secundarios agudos y a largo plazo de estos fármacos (irritabilidad gastrointestinal, inmunodepresión, inhibición corticosuprarrenal, características cushingoides y retraso del crecimiento).
Los corticoesteroides (prednisona y metilprednisolona) se usan cuando la terapia con betabloqueantes está contraindicada o como tratamiento inicial mientras el paciente comienza a recibir el betabloqueante.[125]
Terapia con betabloqueantes tópicos
Los betabloqueantes tópicos se usan sobre todo para el tratamiento de hemangiomas superficiales, localizados y pequeños como alternativa a la observación. También se han usado en combinación con terapia sistémica para los hemangiomas complicados o con el fin de prevenir la reactivación en hemangiomas cuando se comienza a disminuir la terapia sistémica.[126,127,128] Para los betabloqueantes tópicos, se deben seguir las mismas precauciones (evaluación de comorbilidades e historia familiar) que se indicaron antes para el propranolol. La absorción sistémica (plasma y orina) del timolol es variable por lo que es esencial hacer exámenes antes de comenzar el tratamiento para detectar problemas cardíacos, pulmonares y endocrinos. También es importante revisar los antecedentes médicos y exámenes físicos previos. Se debe tener cautela al administrar este fármaco para los hemangiomas ulcerados y profundos ya que es posible observar concentraciones altas de timolol en plasma.[129,130]
Se ha usado timolol tópico en una solución oftálmica de gel al 0,5 %. Se aplica 1 gota en el hemangioma 2 veces por día hasta que se logra una respuesta estable.
Este tratamiento tiene pocos efectos secundarios, pero los lactantes con edad posmenstrual menor de 44 semanas y peso de menos de 2500 g en el momento de iniciar el tratamiento, quizás estén en riesgo de presentar efectos adversos, como bradicardia, hipotensión, apnea e hipotermia.[130,131] La vigilancia minuciosa de la temperatura, la presión arterial y la frecuencia cardíaca al comienzo y durante la terapia con timolol tópico es necesaria en los lactantes prematuros y de bajo peso al nacer, con hemangiomas infantiles.
Evidencia (terapia con timolol tópico):
- En un estudio multicéntrico retrospectivo de cohorte, se trató con timolol al 0,5 % 2 veces por día a 731 niños con hemangiomas de predominio superficial.[128]
- El 92 % de los pacientes mostró una mejora significativa en el color del hemangioma.
- El 72 % de los pacientes mostró una mejora en el tamaño, extensión y volumen del hemangioma.
- Por lo general, el timolol tópico se tolera bien, sin embargo, los datos sobre su inocuidad son escasos.
- Un consorcio español, llevó a cabo un ensayo prospectivo aleatorizado para evaluar la eficacia e inocuidad del timolol tópico para el tratamiento del hemangioma infantil en estadio proliferativo temprano.[132] En este ensayo clínico piloto de fase IIa controlado con placebo, aleatorizado, con enmascaramiento doble multicéntrico participaron pacientes de 10 a 60 días de edad con hemangiomas focales o segmentarios (superficiales, profundos, mixtos o de crecimiento mínimo o interrumpido). Los pacientes se asignaron al azar para recibir una solución tópica de maleato de timolol al 0,5 % o placebo, 2 veces al día por 24 semanas.
- Al cabo de 24 semanas, no hubo diferencias significativas entre el tratamiento con timolol y el placebo en la resolución completa o casi completa del hemangioma infantil (42 % con timolol [n = 11] vs. 36 % con placebo [n = 11]; P = 0,37).
Terapia combinada para los hemangiomas complicados
La terapia combinada se considera al inicio del tratamiento para las lesiones complicadas con alteraciones funcionales o compromiso orgánico, o se utiliza al final de la terapia sistémica para prevenir la reactivación del hemangioma. Se necesita más investigación sobre la eficacia e inocuidad de estos regímenes.
Evidencia (terapia combinada para los hemangiomas complicados):
- En un estudio aleatorizado prospectivo, en el que se comparó el propranolol con 2 semanas de terapia con corticoesteroides y el propranolol solo, se revelaron los siguientes hallazgos:[133]
- Disminución de los tamaños de los hemangiomas al cabo de 2, 4 y 8 semanas en el grupo de terapia combinada, pero sin diferencia estadística de los tamaños a los 6 meses.
- En un estudio aleatorizado prospectivo, en el que se comparó el timolol con propranolol y el propranolol solo, se revelaron los siguientes hallazgos:[134]
Opciones de tratamiento en evaluación clínica para los hemangiomas infantiles
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
El Hemangioma Investigator Group estudia la administración del propranolol en los pacientes de riesgo bajo y estándar mediante consultas virtuales, como consecuencia de la pandemia de COVID-19.[137]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Hemangiomas congénitos
Características clínicas y evaluación diagnóstica
A veces los hemangiomas congénitos son difíciles de diagnosticar, en especial, para los médicos que no están familiarizados con estas lesiones. Los criterios diagnósticos incluyen una lesión purpúrica formada por completo en el momento del nacimiento, con frecuencia hay un halo alrededor de la lesión y se observa flujo alto en la imagen ecográfica. Para el diagnóstico es esencial la observación en serie con el fin de detectar una disminución o al menos una estabilización del tamaño con el paso del tiempo. Estas lesiones no aumentan de tamaño a menos que haya hemorragia dentro del tumor.
Los hemangiomas congénitos se subdividen en las siguientes 3 formas:
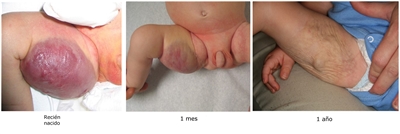
- Hemangiomas congénitos de involución rápida (RICH). Estas lesiones tienen flujo alto y gran tamaño, están formadas en su totalidad al nacer, pero involucionan rápido alrededor de los 12 a 15 meses. Es posible que se ulceren y sangren, y pueden causar insuficiencia cardíaca transitoria y coagulopatía leve. Después de la involución, por lo general se observan algunos cambios residuales en la piel (consultar la Figura 5).[138,139,140,141]
En una serie de casos retrospectivos de hemangiomas congénitos, se observaron varios hallazgos ecográficos de riesgo alto para RICH. Los lagos venosos se relacionaron con insuficiencia cardíaca y se observó un mayor riesgo de hemorragia con los lagos venosos y la ectasia venosa. Los lactantes con RICH se deben evaluar mediante ecografía y vigilancia rigurosa si se observan estas características de riesgo alto.[142]
- Hemangiomas congénitos de involución parcial (PICH). Estas lesiones están formadas en su totalidad al nacer y su involución es parcial.[143]
- Hemangiomas congénitos sin involución (NICH). Estas lesiones están formadas al nacer y nunca involucionan. Según la localización de las lesiones y si causan deterioro funcional, es posible que se necesite extirpación quirúrgica.[144,145]
Características moleculares e histopatológicas
Los hemangiomas congénitos son tumores vasculares benignos que proliferan in utero. La evolución de estas lesiones es total al momento de nacer. En el estudio histológico estas lesiones son negativas para GLUT1, a diferencia de los hemangiomas infantiles. Estos hemangiomas suelen ser cutáneos, pero a veces se localizan en las vísceras. Las complicaciones son hemorragia, insuficiencia cardíaca transitoria y coagulopatía transitoria.[146]
Se encontraron algunas variantes somáticas activadoras de GNAQ y GNA11 que se relacionan con los hemangiomas congénitos.[147] Se necesitan estudios adicionales para evaluar la importancia de estos hallazgos, ya que es posible que ayuden al diagnóstico y a entender las características fisiopatológicas.
Tumores vasculares hepáticos
A partir de la creación de las nuevas clasificaciones de la OMS y la ISSVA, ha cambiado la terminología de los tumores vasculares hepáticos pediátricos.[148,149,150] La clasificación histórica de Dehner de los tipos 1, 2 y 3 de hemangioendotelioma hepático ya no es la preferida por los patólogos.[151] El término hemangioendotelioma no se considera una entidad aislada.
En las IRM, los tumores vasculares hepáticos son hiperintensos en las imágenes T2 e hipotensos en las imágenes T1; las imágenes después del contraste exhiben realce periférico temprano con realce difuso posterior.[76] En la práctica médica, estos tumores se han clasificado de acuerdo con sus características clínicas y la evaluación radiológica.[76,152]
Las lesiones se suelen dividir en las siguientes 3 categorías:[76,152]
En una clasificación más adecuada se usa una evaluación interdisciplinar, que incluye la clasificación patológica con evaluación genómica, la evaluación radiológica por imagen, así como los antecedentes y exámenes clínicos; esto se fundamenta en las clasificaciones de la ISSVA y la OMS. En un estudio se analizaron 33 casos de tumores vasculares hepáticos pediátricos a partir de sus características anatomoclínicas desde 1970 hasta 2021.[153] De los casos, 13 se identificaron como hemangiomas congénitos hepáticos; todas eran lesiones únicas, y la mayoría de ellas eran RICH. De los pacientes, 10 tenían hemangiomas infantiles hepáticos; 3 presentaban angiosarcoma hepático; y 1, hemangioendotelioma epitelioide hepático. Se excluyó del estudio a 6 pacientes, 5 con malformaciones vasculares y 1 con hamartoma mesenquimal vascular dominante. El estudio reveló la importancia de un abordaje de equipo interdisciplinario en la evaluación de estos tumores.
Hemangiomas congénitos
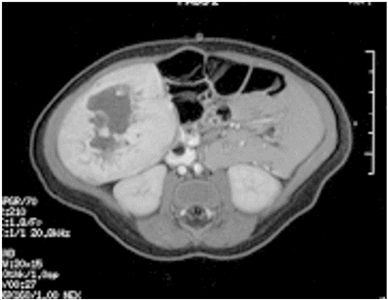
Las lesiones hepáticas focales, por lo general son hemangiomas congénitos (RICH o NICH, con poca frecuencia PICH) (consultar la Figura 6). Es posible que los RICH se manifiesten con síntomas de insuficiencia cardíaca y coagulopatía leve a moderada, pero por lo habitual, se detectan en una ecografía prenatal o con la presencia de una masa asintomática en el periodo neonatal.
Las opciones de tratamiento para las lesiones vasculares hepáticas focales son las siguientes:
- Tratamiento de apoyo. La mayoría de estas lesiones son asintomáticas y es posible vigilarlas durante la involución mediante ecografía.
- Embolización. Este procedimiento se tiene en cuenta en el caso de derivaciones sintomáticas graves que no responden al tratamiento para la insuficiencia cardíaca congestiva. Este procedimiento lo debe realizar un radiólogo intervencionista con pericia en el tratamiento de anomalías vasculares.[154]
- Cirugía. Los pacientes con un hemangioma congénito de hígado focal sintomático grande que no responde al tratamiento de apoyo o a la intervención radiológica quizás sean aptos para someterse a una resección quirúrgica. Esta es una situación infrecuente que debe evaluar un equipo interdisciplinario de anomalías vasculares. Las indicaciones para la extirpación quirúrgica incluyen ruptura, hemorragia y coagulopatía sin resolver. Se informó sobre 2 pacientes que necesitaron resección quirúrgica después de presentar ascitis clínicamente significativa durante la involución de RICH.[155,156]
Ningún medicamento ha demostrado ser un tratamiento eficaz para estas lesiones; los lactantes necesitan apoyo durante este período inicial hasta que comienza la involución.[76,152] Es posible diagnosticar estas lesiones antes del nacimiento. En situaciones poco frecuentes, el tratamiento materno con medicamentos como corticoesteroides fue eficaz, pero es más probable que la causa haya sido una involución natural.[157]
Hemangiomas infantiles
Las lesiones hepáticas multifocales son hemangiomas infantiles. Es posible que no sea necesario tratar las lesiones multifocales si el paciente es asintomático; por lo general, estas lesiones siguen el mismo curso proliferativo y de involución que los hemangiomas cutáneos.[76,152] Estas lesiones se vigilan de cerca y, si hay crecimiento, se debe considerar la terapia con propranolol. Si se necesita propranolol, las dosis de hasta 2 mg/kg por día son eficaces.
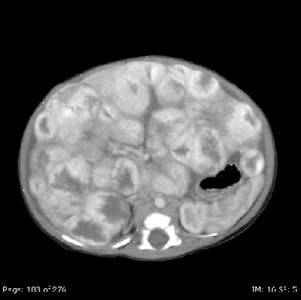
Hemangiomas hepáticos infantiles difusos
Las lesiones hepáticas difusas son muy graves (consultar la Figura 7). Las complicaciones incluyen hipotiroidismo causado por expresión de la yodotironina desyodinasa, insuficiencia cardíaca congestiva o de alto gasto y síndrome compartimental abdominal.[75,76,158,159]
Las opciones de tratamiento de las lesiones hepáticas difusas son las siguientes:
- Propranolol: los betabloqueantes constituyen el tratamiento más común para los hemangiomas infantiles difusos de hígado y algunos hemangiomas multifocales de hígado. Se indican dosis de tratamiento de 2 mg/kg a 3 mg/kg por día.[92]
- Remplazo de hormona tiroidea: la terapia de remplazo de hormona tiroidea debe ser intensiva si se diagnostica hipotiroidismo. Es posible que se necesite un tratamiento con dosis más altas de hormonas debido al consumo intenso de estas por parte del tumor, lo que causa la deficiencia.[78]
- Quimioterapia: se han utilizado corticoesteroides, ciclofosfamida y vincristina para tratar el hemangioma hepático difuso infantil.[76,160,161]
- Trasplante: si un paciente no responde bien al tratamiento médico, es posible que esté indicado realizar un trasplante.[162] El trasplante se considera solo para los pacientes con lesiones difusas graves que tienen insuficiencia orgánica multisistémica y no hay tiempo suficiente para usar una farmacoterapia eficaz.
Se notificaron casos aislados de neoplasia maligna en pacientes con hemangiomas infantiles hepáticos difusos.[163,164] No está claro que todos los casos se debieran a una transformación de una lesión benigna a un fenotipo maligno. Sin embargo, si la lesión no responde bien a la terapia estándar se debe considerar una biopsia. Se necesitan más evaluación y consenso para determinar si estos pacientes se deben vigilar durante un período de tiempo más largo con ecografía hepática. Para obtener más información, consultar la sección Angiosarcoma.
El diagnóstico diferencial de las lesiones vasculares hepáticas siempre incluye los tumores hepáticos malignos; por lo tanto, la alfafetoproteína (AFP) se debe incluir en los análisis de laboratorio iniciales. Las concentraciones de AFP son muy altas en todos los recién nacidos, pero disminuyen, en forma rápida, a una concentración normal al cabo de varios meses. Estas concentraciones deben disminuir rápido, pero de no ser así o si tienden a aumentar se debe sospechar un hepatoblastoma. No hay estudios prospectivos en los que se investigue las concentraciones altas de AFP, en pacientes con hemangiomas.[165,166] En recién nacidos con insuficiencia cardíaca congestiva, algunos hepatoblastomas hipervasculares se han diagnosticado por error como hemangiomas infantiles. Otros tumores de diagnóstico diferencial son los angiosarcomas, los neuroblastomas metastásicos y los hamartomas mesenquimatosos. Si hay dudas sobre el diagnóstico, se recomienda una biopsia, aunque hay riesgo de hemorragia por el procedimiento.[167]
| Características | Hemangioma congénito hepático (HCH) | Hemangioma infantil hepático (HIH) | Angiosarcoma hepático (AH) | Hemangioendotelio epitelioide hepático (HEHE) |
|---|---|---|---|---|
| ICC = insuficiencia cardíaca congestiva; NICH = hemangioma congénito sin involución; PICH = hemangioma congénito de involución parcial; RICH = hemangioma congénito de involución rápida. | ||||
| a Adaptación de Berklite et al.[153] | ||||
| Cuadro clínico inicial | Se observa en el momento del nacimiento, o antes; ICC; coagulopatía transitoria; lesiones únicas; RICH, PICH, con poca frecuencia NICH | Se observa después del nacimiento, por lo general asociado a lesiones cutáneas; lesiones difusas con hipotiroidismo significativo e ICC | Poco frecuente en pediatría, se ha observado en neonatos y niños pequeños; muy maligno | Muy poco frecuente; asociado a otras lesiones (hueso, pulmón); curso variable |
| Imágenes | Lesión sólida | Lesiones múltiples o difusas | Grandes lesiones infiltrativas, pueden ser difusas | Lesiones sólidas o múltiples |
| Características histológicas | Cambios involutivos (calcificación, necrosis), vasos capilares del estroma dilatados y fibróticos | Vasculatura sinusoidal anastomosada, células endoteliales densas de aspecto normal | Atipia citológica marcada, células tumorales infiltrantes, epitelioides a fusiformes, marcada actividad mitótica | Células endoteliales epitelioides en un fondo de estroma mixohialino |
| GLUT1 | Negativo | Positivo | Positivo en el 20 % de los tumores | Negativo |
| Variantes somáticas o fusiones génicas | GNAQ,GNA11 | Ninguno | KRAS,KDR,PTPRB,FLT4,PLCG1,PIK3CA,TP53,TIE1,AKT1,CIC | YAP1::TFE3,WWTR1::CAMTA1 |
Hemangioma fusocelular
Cuadro clínico, características moleculares e histopatológicas
Los hemangiomas fusocelulares, en un comienzo llamados hemangioendoteliomas fusocelulares, a menudo se presentan como lesiones superficiales (piel y tejido subcutáneo) y dolorosas que comprometen las extremidades distales en niños y adultos.[168,169] Los tumores se ven como lesiones de color marrón rojizo o azulado que comienzan como un solo nódulo y luego, con el paso de los años, se convierten en lesiones multifocales dolorosas. Los hemangiomas son bien circunscritos, a veces contienen flebolitos y tienen espacios cavernosos llenos de sangre que se alternan con áreas nodulares de proliferación de células fusiformes. Un porcentaje importante de hemangiomas fusocelulares son intravasculares por completo. La vena que contiene el tumor es anormal, así como los vasos sanguíneos que no forman parte de la masa tumoral.[170,171]
Es posible ver los hemangiomas fusocelulares en pacientes con síndrome de Maffuci (hemangiomas cutáneos de células fusiformes que se presentan con tumores cartilaginosos y encondromas), el síndrome de Klippel-Trenaunay (malformaciones capilares, linfáticas o venosas), las anomalías linfáticas generalizadas, el linfedema y el trombo organizado.[170,171] En el síndrome de Maffuci, los hemangiomas de células fusiformes se relacionan con variantes de IDH1 o IDH2.[172]
Tratamiento del hemangioma fusocelular
No hay un tratamiento estándar para el hemangioma fusocelular porque no se ha estudiado en ensayos clínicos. La extirpación quirúrgica suele ser curativa, aunque hay un riesgo de recidiva.[170,171]
Hemangioma epitelioide
Cuadro clínico y características histopatológicas
Los hemangiomas epitelioides (HE) son lesiones benignas que, por lo general, se presentan en la piel y la hipodermis, pero también se presentan en otras áreas, como el hueso, con lesiones focales o multifocales.[170,173] Es posible que los hemangiomas epitelioides sean un proceso reactivo porque se pueden relacionar con un trauma local y surgir durante el embarazo. Los pacientes suelen presentar inflamación local y dolor en el sitio comprometido. En el hueso, se presentan como lesiones líticas bien definidas que comprometen la metáfisis y la diáfisis de los huesos largos.[170,174] Pueden tener un modelo mixto de destrucción ósea lítica y esclerótica.
En la evaluación patológica, los hemangiomas epitelioides exhiben capilares de calibre pequeño con citoplasma eosinofílico y vacuolado, y núcleos ovales grandes con surcos y lobulados. Las células endoteliales son células hinchadas y maduras, y se observan vasos bien formados rodeados de múltiples células endoteliales epitelioides con citoplasma abundante. Carecen de atipia celular y actividad mitótica.[170,173,174,175]
En un estudio de 58 casos de hemangiomas epitelioides, se encontraron reordenamientos del gen FOS en el 29 % de estos. Los reordenamientos del gen FOS se observaron con mayor frecuencia en los hemangiomas epitelioides celulares y en las lesiones intraóseas que en las lesiones de piel, tejido blando, cabeza y cuello. Esta anomalía genética quizás sea útil al momento de distinguir los hemangiomas epitelioides de otros tumores vasculares epitelioides malignos.[175]
En un informe de 1 sola institución, se revisaron 11 pacientes con hemangiomas epitelioides (mediana de edad, 14,4 años) diagnosticados entre 1999 y 2017. Las lesiones se presentaron en las extremidades inferiores (5 pacientes), el cráneo (3 pacientes), la pelvis (2 pacientes) y la columna (1 paciente). En 5 pacientes, se observó enfermedad multifocal. Los pacientes presentaron dolor localizado y síntomas neurológicos, incluso daño de pares craneales. No se observó atipia citológica significativa y las células endoteliales fueron positivas para CD31 y ERG, y negativas para citoqueratina y CAMPTA1. La mediana de seguimiento fue de 1,5 años. Se usaron varias modalidades de tratamiento, como la cirugía, la embolización endovascular, la crioablación y el tratamiento médico. De los pacientes, 1 recibió sirólimus y otro paciente recibió interferón; en ambos casos las lesiones se redujeron durante el primer año de seguimiento. El paciente más joven, de 2,5 años, presentó lesiones craneales multifocales que remitieron de forma parcial 1 año después sin tratamiento.[176]
Tratamiento del hemangioma epitelioide
No hay un tratamiento estándar para el hemangioma epitelioide porque no se ha estudiado en ensayos clínicos. El tratamiento a menudo consiste en curetaje, escleroterapia o resección. En casos poco frecuentes, se utiliza radioterapia.[170,174]
Granuloma piógeno (hemangioma capilar lobulillar)
Cuadro clínico inicial, características histopatológicas y moleculares
Los granulomas piógenos (PG), que se conocen como hemangiomas capilares lobulillares, son lesiones reactivas benignas. Los granulomas piógenos se presentan en cualquier edad, incluso al nacer (congénitos), durante el período neonatal, la lactancia o el embarazo aunque son más comunes en niños mayores y adultos jóvenes. Estas lesiones pueden surgir de manera espontánea, en sitios de trauma o dentro de malformaciones capilares o arteriovenosas. Los granulomas piógenos también se relacionaron con medicamentos como los anticonceptivos orales y los retinoides.
Se presentan como masas solitarias, pero se han descrito lesiones múltiples (agrupadas) o, de manera infrecuente, lesiones diseminadas.[177] Estas lesiones se ven como nódulos vasculares pequeños o grandes, lisos o lobulados, que en ocasiones crecen rápido, a veces durante semanas o meses, y que tienden a sangrar de manera profusa. Por lo general, estas lesiones son cutáneas, pero se han notificado granulomas piógenos profundos o subcutáneos que imitan otras lesiones vasculares.[178] Desde el punto de vista histológico, estas lesiones se componen de capilares y vénulas con células endoteliales hinchadas separadas en lóbulos por estroma fibromixoide. Con el tiempo, algunas lesiones no tratadas se atrofian, se tornan fibromatosas y remiten en forma lenta. En una revisión retrospectiva de una serie de 8 niños con granulomas piógenos diseminados congénitos o neonatales, se notificó lesiones hemorrágicas en el sistema nervioso central en 7 pacientes, 5 de los cuales presentaron secuelas neurológicas. De los 8 pacientes, 4 presentaron coagulopatía transitoria.[179][Nivel de evidencia C2]
Se desconoce la patogenia de los granulomas piógenos relacionados con malformaciones capilares y de los esporádicos. En un estudio se investigó a 10 pacientes con granulomas piógenos derivados de una malformación capilar. Se detectó en 8 pacientes variantes BRAF c.1799T>A, en 1 paciente una variante NRAS c.2A>G y en 1 paciente una variante GNAQ c.548G>A. También se detectó la variante de GNAQ en la malformación capilar subyacente. De 25 pacientes con granulomas piógenos y sin malformaciones capilares, 3 tuvieron variantes BRAF c.1799T>A y 1 paciente tuvo una variante KRAS c.37G>C. Estos hallazgos genéticos contribuirán a las modalidades de tratamiento futuras para este tumor vascular benigno.[180]
Tratamiento del granuloma piógeno
La escisión de espesor total es el tratamiento con la tasa de recidiva más baja (cerca del 3 %),[181] pero también es posible usar el curetaje, la fotocoagulación con láser o la crioterapia.[182] También se han utilizado timolol tópico y propranolol.
Evidencia (betabloqueantes tópicos):
- En una serie de un solo grupo de pacientes con granulomas piógenos adquiridos en el ojo, una cantidad pequeña de pacientes recibió timolol tópico al 0,5 %, 2 veces al día durante un período de 21 días a 6 semanas.[183,184][Nivel de evidencia C3]
- Se observaron respuestas completas o casi completas sin recidivas subsecuentes ni progresión en el 75 % al 100 % de los pacientes (todas las edades).
- En un estudio de 22 pacientes con granulomas piógenos cutáneos, tratados con ungüento de propranolol tópico al 1 % con oclusión, se obtuvo los siguientes resultados:[185]
- El 59 % de los pacientes logró una respuesta completa (media, 66 días), el 18 % de los pacientes tuvo enfermedad estable y el 22 % de los pacientes no respondió al tratamiento.
- En este estudio, solo se evaluaron los efectos tóxicos en la piel.
- Los autores no hicieron comentarios sobre la penetrancia de la formulación de propranolol ni incluyeron una evaluación de la inocuidad de los efectos secundarios, como la hipoglucemia, ni sobre los efectos en la frecuencia cardíaca o la presión arterial.
Angiofibroma
Cuadro clínico inicial
Los angiofibromas son neoplasias benignas poco frecuentes en la población pediátrica. Por lo general, son lesiones cutáneas relacionadas con esclerosis tuberosa, que se ven como pápulas rojas en la cara.
Tratamiento del angiofibroma
Se han utilizado la escisión del tumor, los tratamientos con láser y los tratamientos tópicos como el sirólimus.[186,187,188]
Evidencia (sirólimus tópico):
- En un ensayo prospectivo, controlado con placebo, aleatorizado de 9 centros de oncología en los que participaron 62 pacientes que recibieron sirólimus en gel, se observó los siguientes resultados:[189]
- El 60 % de los pacientes que recibieron sirólimus, mostró una mejora importante en el tamaño y el color de la lesión, la cual se evaluó en la semana 12.
- En otro estudio prospectivo, multicéntrico, aleatorizado, con enmascaramiento doble y controlado con vehículo que incluyó 6 visitas mensuales a la clínica, 179 pacientes con angiofibromas faciales relacionados con esclerosis tuberosa compleja recibieron tratamiento con rapamicina (sirólimus) tópica 0,3 g cada 30 g (1 %).[190]
- Según la Angiofibroma Grading Scale, los pacientes que recibieron tratamiento con rapamicina tópica mostraron una mejora estadística y clínicamente significativa de los angiofibromas faciales.
Angiofibroma nasofaríngeo juvenil
Cuadro clínico inicial y características histopatológicas
Los angiofribromas nasofaríngeos juveniles representan el 0,5 % de todos los tumores de cabeza y cuello.[191] Por lo general se presentan en varones peripubertales. Si bien no es típico que los angiofribromas nasofaríngeos juveniles se incluyan entre los tumores vasculares, desde el punto de vista histológico, estos tumores son tumores vasculares, con células que expresan marcadores vasculares endoteliales CD31, VEGFA y VEGFR1.
Pese a sus características histológicas benignas, es posible que los angiofibromas nasofaríngeos juveniles sean destructivos a nivel local, al diseminarse desde la cavidad nasal a la nasofaringe, los senos paranasales, la órbita y la base del cráneo con extensión intracraneal. En algunas publicaciones se indicó una influencia hormonal en los angiofibromas nasofaríngeos juveniles, con énfasis en los mecanismos moleculares participantes.[192,193] Se sometieron a tomografía por emisión de positrones-tomografía computarizada con galio Ga 68-[DOTA, 1-Nal3]-octreotida (68Ga-DOTANOC) 19 pacientes con angiofibromas nasofaríngeos juveniles primarios diagnosticados de forma radiológica y clínica.[194] Se usó está prueba debido a la alta expresión de receptores de somatostatina (SSTR) en estos tumores. Se observó expresión de DOTANOC en todos los 19 casos de tumores nasofaríngeos juveniles primarios (100 %). La media del cociente del valor máximo de captación estandarizado de DOTANOC del tumor y la actividad de fondo fue de 6,9 (±1,4) (intervalo, 3,8–9,5). Se visualizó de forma notoria, una extensión intracraneal en 13 de 19 pacientes debido a la falta de captación de DOTANOC del encéfalo. Los autores sugieren que estos hallazgos ofrecen posibilidades para la imaginología diagnóstica fisiológica, ya que prometen una mayor especificidad y sensibilidad. Es posible que este tipo de imagen se use en situaciones diagnósticas ambivalentes, como la detección de una recidiva.
Tratamiento del angiofibroma nasofaríngeo juvenil
Si bien la escisión quirúrgica es el tratamiento preferido, quizás sea difícil debido a la extensión de la lesión. En una revisión retrospectiva de una sola institución sobre los angiofibromas nasofaríngeos juveniles, se identificó a 37 pacientes con diseminación lateral.[195] La diseminación anterolateral a la fosa pterigopalatina se presentó en 36 pacientes (97 %) y la diseminación más allá de la fosa infratemporal en 20 pacientes (54 %). En 16 pacientes (43 %), se observó diseminación posterolateral (posterior a la apófisis pterigoidea o entre sus placas). La tasa de recidiva fue del 29,7 % (11 de 37 pacientes). La tasa de recidiva en pacientes con diseminación anterior o posterolateral fue significativamente más alta que en los pacientes con diseminación anterolateral sola.
Los angiofibromas nasofaríngeos juveniles también se han tratado con radioterapia, quimioterapia, terapia con interferón α y sirólimus.[196,197,198,199,200]
Referencias:
- Kilcline C, Frieden IJ: Infantile hemangiomas: how common are they? A systematic review of the medical literature. Pediatr Dermatol 25 (2): 168-73, 2008 Mar-Apr.
- Munden A, Butschek R, Tom WL, et al.: Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol 170 (4): 907-13, 2014.
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma. Pediatrics 136 (4): e1060-104, 2015.
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma: Executive Summary. Pediatrics 136 (4): 786-91, 2015.
- Haggstrom AN, Drolet BA, Baselga E, et al.: Prospective study of infantile hemangiomas: demographic, prenatal, and perinatal characteristics. J Pediatr 150 (3): 291-4, 2007.
- Dickison P, Christou E, Wargon O: A prospective study of infantile hemangiomas with a focus on incidence and risk factors. Pediatr Dermatol 28 (6): 663-669, 2011.
- Chen XD, Ma G, Chen H, et al.: Maternal and perinatal risk factors for infantile hemangioma: a case-control study. Pediatr Dermatol 30 (4): 457-61, 2013.
- Chang LC, Haggstrom AN, Drolet BA, et al.: Growth characteristics of infantile hemangiomas: implications for management. Pediatrics 122 (2): 360-7, 2008.
- Tollefson MM, Frieden IJ: Early growth of infantile hemangiomas: what parents' photographs tell us. Pediatrics 130 (2): e314-20, 2012.
- Haggstrom AN, Lammer EJ, Schneider RA, et al.: Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics 117 (3): 698-703, 2006.
- Waner M, North PE, Scherer KA, et al.: The nonrandom distribution of facial hemangiomas. Arch Dermatol 139 (7): 869-75, 2003.
- Chiller KG, Passaro D, Frieden IJ: Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol 138 (12): 1567-76, 2002.
- Metry DW, Garzon MC, Drolet BA, et al.: PHACE syndrome: current knowledge, future directions. Pediatr Dermatol 26 (4): 381-98, 2009 Jul-Aug.
- Munabi NC, Tan QK, Garzon MC, et al.: Growth Hormone Induces Recurrence of Infantile Hemangiomas After Apparent Involution: Evidence of Growth Hormone Receptors in Infantile Hemangioma. Pediatr Dermatol 32 (4): 539-43, 2015 Jul-Aug.
- Olsen GM, Nackers A, Drolet BA: Infantile and congenital hemangiomas. Semin Pediatr Surg 29 (5): 150969, 2020.
- Haggstrom AN, Drolet BA, Baselga E, et al.: Prospective study of infantile hemangiomas: clinical characteristics predicting complications and treatment. Pediatrics 118 (3): 882-7, 2006.
- Braun M, Metry D, Frieden IJ, et al.: Persistent dysesthesias in involuted infantile hemangiomas: An uncommon complication in a common condition. Pediatr Dermatol 38 (5): 1061-1065, 2021.
- Baselga E, Roe E, Coulie J, et al.: Risk Factors for Degree and Type of Sequelae After Involution of Untreated Hemangiomas of Infancy. JAMA Dermatol 152 (11): 1239-1243, 2016.
- Blei F, Walter J, Orlow SJ, et al.: Familial segregation of hemangiomas and vascular malformations as an autosomal dominant trait. Arch Dermatol 134 (6): 718-22, 1998.
- Castrén E, Salminen P, Vikkula M, et al.: Inheritance Patterns of Infantile Hemangioma. Pediatrics 138 (5): , 2016.
- Greenberger S, Bischoff J: Pathogenesis of infantile haemangioma. Br J Dermatol 169 (1): 12-9, 2013.
- Biswas A, Richards JE, Massaro J, et al.: Mast cells in cutaneous tumors: innocent bystander or maestro conductor? Int J Dermatol 53 (7): 806-11, 2014.
- Khan ZA, Boscolo E, Picard A, et al.: Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest 118 (7): 2592-9, 2008.
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001.
- Yu Y, Flint AF, Mulliken JB, et al.: Endothelial progenitor cells in infantile hemangioma. Blood 103 (4): 1373-5, 2004.
- Boscolo E, Mulliken JB, Bischoff J: Pericytes from infantile hemangioma display proangiogenic properties and dysregulated angiopoietin-1. Arterioscler Thromb Vasc Biol 33 (3): 501-9, 2013.
- Zhang H, Wei T, Johnson A, et al.: NOTCH pathway activation in infantile hemangiomas. J Vasc Surg Venous Lymphat Disord 9 (2): 489-496, 2021.
- Barnés CM, Huang S, Kaipainen A, et al.: Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A 102 (52): 19097-102, 2005.
- Walter JW, North PE, Waner M, et al.: Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer 33 (3): 295-303, 2002.
- Ritter MR, Dorrell MI, Edmonds J, et al.: Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A 99 (11): 7455-60, 2002.
- Takahashi K, Mulliken JB, Kozakewich HP, et al.: Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 93 (6): 2357-64, 1994.
- Ritter MR, Reinisch J, Friedlander SF, et al.: Myeloid cells in infantile hemangioma. Am J Pathol 168 (2): 621-8, 2006.
- Bielenberg DR, Bucana CD, Sanchez R, et al.: Progressive growth of infantile cutaneous hemangiomas is directly correlated with hyperplasia and angiogenesis of adjacent epidermis and inversely correlated with expression of the endogenous angiogenesis inhibitor, IFN-beta. Int J Oncol 14 (3): 401-8, 1999.
- Colonna V, Resta L, Napoli A, et al.: Placental hypoxia and neonatal haemangioma: clinical and histological observations. Br J Dermatol 162 (1): 208-9, 2010.
- de Jong S, Itinteang T, Withers AH, et al.: Does hypoxia play a role in infantile hemangioma? Arch Dermatol Res 308 (4): 219-27, 2016.
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001.
- Dubois J, Patriquin HB, Garel L, et al.: Soft-tissue hemangiomas in infants and children: diagnosis using Doppler sonography. AJR Am J Roentgenol 171 (1): 247-52, 1998.
- Horii KA, Drolet BA, Frieden IJ, et al.: Prospective study of the frequency of hepatic hemangiomas in infants with multiple cutaneous infantile hemangiomas. Pediatr Dermatol 28 (3): 245-53, 2011 May-Jun.
- Ma EH, Robertson SJ, Chow CW, et al.: Infantile Hemangioma with Minimal or Arrested Growth: Further Observations on Clinical and Histopathologic Findings of this Unique but Underrecognized Entity. Pediatr Dermatol 34 (1): 64-71, 2017.
- Valdivielso-Ramos M, Torrelo A, Martin-Santiago A, et al.: Infantile hemangioma with minimal or arrested growth as the skin manifestation of PHACE syndrome. Pediatr Dermatol 35 (5): 622-627, 2018.
- Planas-Ciudad S, Roé Crespo E, Sánchez-Carpintero I, et al.: Infantile hemangiomas with minimal or arrested growth associated with soft tissue hypertrophy: a case series of 10 patients. J Eur Acad Dermatol Venereol 31 (11): 1924-1929, 2017.
- Elluru RG, Friess MR, Richter GT, et al.: Multicenter Evaluation of the Effectiveness of Systemic Propranolol in the Treatment of Airway Hemangiomas. Otolaryngol Head Neck Surg 153 (3): 452-60, 2015.
- McCormick AA, Tarchichi T, Azbell C, et al.: Subglottic hemangioma: Understanding the association with facial segmental hemangioma in a beard distribution. Int J Pediatr Otorhinolaryngol 113: 34-37, 2018.
- Xue L, Sun C, Xu DP, et al.: Clinical Outcomes of Infants With Periorbital Hemangiomas Treated With Oral Propranolol. J Oral Maxillofac Surg 74 (11): 2193-2199, 2016.
- Proisy M, Powell J, McCuaig C, et al.: PHACES Syndrome and Associated Anomalies: Risk Associated With Small and Large Facial Hemangiomas. AJR Am J Roentgenol 217 (2): 507-514, 2021.
- Theiler M, Baselga E, Gerth-Kahlert C, et al.: Infantile hemangiomas with conjunctival involvement: An underreported occurrence. Pediatr Dermatol 34 (6): 681-685, 2017.
- Garzon MC, Epstein LG, Heyer GL, et al.: PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr 178: 24-33.e2, 2016.
- Frieden IJ, Reese V, Cohen D: PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol 132 (3): 307-11, 1996.
- Metry D, Heyer G, Hess C, et al.: Consensus Statement on Diagnostic Criteria for PHACE Syndrome. Pediatrics 124 (5): 1447-56, 2009.
- Metry DW, Haggstrom AN, Drolet BA, et al.: A prospective study of PHACE syndrome in infantile hemangiomas: demographic features, clinical findings, and complications. Am J Med Genet A 140 (9): 975-86, 2006.
- Drolet BA, Dohil M, Golomb MR, et al.: Early stroke and cerebral vasculopathy in children with facial hemangiomas and PHACE association. Pediatrics 117 (3): 959-64, 2006.
- Heyer GL, Dowling MM, Licht DJ, et al.: The cerebral vasculopathy of PHACES syndrome. Stroke 39 (2): 308-16, 2008.
- Haggstrom AN, Garzon MC, Baselga E, et al.: Risk for PHACE syndrome in infants with large facial hemangiomas. Pediatrics 126 (2): e418-26, 2010.
- Poindexter G, Metry DW, Barkovich AJ, et al.: PHACE syndrome with intracerebral hemangiomas, heterotopia, and endocrine dysfunction. Pediatr Neurol 36 (6): 402-6, 2007.
- Brandon K, Burrows P, Hess C, et al.: Arteriovenous malformation: a rare manifestation of PHACE syndrome. Pediatr Dermatol 28 (2): 180-4, 2011 Mar-Apr.
- Chan YC, Eichenfield LF, Malchiodi J, et al.: Small facial haemangioma and supraumbilical raphe--a forme fruste of PHACES syndrome? Br J Dermatol 153 (5): 1053-7, 2005.
- Nabatian AS, Milgraum SS, Hess CP, et al.: PHACE without face? Infantile hemangiomas of the upper body region with minimal or absent facial hemangiomas and associated structural malformations. Pediatr Dermatol 28 (3): 235-41, 2011 May-Jun.
- Antonov NK, Spence-Shishido A, Marathe KS, et al.: Orbital Hemangioma with Intracranial Vascular Anomalies and Hemangiomas: A New Presentation of PHACE Syndrome? Pediatr Dermatol 32 (6): e267-72, 2015 Nov-Dec.
- Burrows PE, Robertson RL, Mulliken JB, et al.: Cerebral vasculopathy and neurologic sequelae in infants with cervicofacial hemangioma: report of eight patients. Radiology 207 (3): 601-7, 1998.
- Hess CP, Fullerton HJ, Metry DW, et al.: Cervical and intracranial arterial anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol 31 (10): 1980-6, 2010.
- Yu J, Siegel DH, Drolet BA, et al.: Prevalence and Clinical Characteristics of Headaches in PHACE Syndrome. J Child Neurol 31 (4): 468-73, 2016.
- Samuelov L, Kinori M, Mancini AJ, et al.: Ocular Complications in PHACE Syndrome: A True Association or a Coincidence? J Pediatr 204: 214-218.e2, 2019.
- Stefanko NS, Davies OMT, Beato MJ, et al.: Hamartomas and midline anomalies in association with infantile hemangiomas, PHACE, and LUMBAR syndromes. Pediatr Dermatol 37 (1): 78-85, 2020.
- Martin KL, Arvedson JC, Bayer ML, et al.: Risk of dysphagia and speech and language delay in PHACE syndrome. Pediatr Dermatol 32 (1): 64-9, 2015 Jan-Feb.
- Chiu YE, Siegel DH, Drolet BA, et al.: Tooth enamel hypoplasia in PHACE syndrome. Pediatr Dermatol 31 (4): 455-8, 2014 Jul-Aug.
- Duffy KJ, Runge-Samuelson C, Bayer ML, et al.: Association of hearing loss with PHACE syndrome. Arch Dermatol 146 (12): 1391-6, 2010.
- Letertre O, Boccara O, Prey S, et al.: Segmental facial infantile haemangiomas in the era of propranolol: evaluation at 6 years of age. J Eur Acad Dermatol Venereol 36 (4): 610-614, 2022.
- Braun M, Frieden IJ, Siegel DH, et al.: Multicenter Study of Long-Term Outcomes and Quality of Life in PHACE Syndrome after Age 10. J Pediatr 267: 113907, 2024.
- Iacobas I, Burrows PE, Frieden IJ, et al.: LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr 157 (5): 795-801.e1-7, 2010.
- Girard C, Bigorre M, Guillot B, et al.: PELVIS Syndrome. Arch Dermatol 142 (7): 884-8, 2006.
- Stockman A, Boralevi F, Taïeb A, et al.: SACRAL syndrome: spinal dysraphism, anogenital, cutaneous, renal and urologic anomalies, associated with an angioma of lumbosacral localization. Dermatology 214 (1): 40-5, 2007.
- Anwar T, Malm-Buatsi E: Propranolol as an effective therapy for infantile haemangioma of the urinary bladder. BMJ Case Rep 12 (2): , 2019.
- Drolet BA, Chamlin SL, Garzon MC, et al.: Prospective study of spinal anomalies in children with infantile hemangiomas of the lumbosacral skin. J Pediatr 157 (5): 789-94, 2010.
- Sardana K, Gupta R, Garg VK, et al.: A prospective study of cutaneous manifestations of spinal dysraphism from India. Pediatr Dermatol 26 (6): 688-95, 2009 Nov-Dec.
- Rialon KL, Murillo R, Fevurly RD, et al.: Risk factors for mortality in patients with multifocal and diffuse hepatic hemangiomas. J Pediatr Surg 50 (5): 837-41, 2015.
- Hsi Dickie B, Fishman SJ, Azizkhan RG: Hepatic vascular tumors. Semin Pediatr Surg 23 (4): 168-72, 2014.
- Ji Y, Chen S, Yang K, et al.: Screening for infantile hepatic hemangioma in patients with cutaneous infantile hemangioma: A multicenter prospective study. J Am Acad Dermatol 84 (5): 1378-1384, 2021.
- Huang SA, Tu HM, Harney JW, et al.: Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 343 (3): 185-9, 2000.
- Fernández Faith E, Shah S, Witman PM, et al.: Clinical Features, Prognostic Factors, and Treatment Interventions for Ulceration in Patients With Infantile Hemangioma. JAMA Dermatol 157 (5): 566-572, 2021.
- de Graaf M, Knol MJ, Totté JE, et al.: E-learning enables parents to assess an infantile hemangioma. J Am Acad Dermatol 70 (5): 893-8, 2014.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, et al.: The Infantile Hemangioma Referral Score: A Validated Tool for Physicians. Pediatrics 145 (4): , 2020.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al.: Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics 143 (1): , 2019.
- Kessels JP, Hamers ET, Ostertag JU: Superficial hemangioma: pulsed dye laser versus wait-and-see. Dermatol Surg 39 (3 Pt 1): 414-21, 2013.
- Trapeznikova TV, Pisklakova TP, Khomchenko VV, et al.: New technology for coagulation of dilated vessels using the combined effects of several modes of generation and wavelengths in one laser pulse for the treatment of pediatric hemangiomas: Open prospective study. Dermatol Ther 33 (3): e13341, 2020.
- He HY, Shi WK, Jiang JC, et al.: An exploration of optimal time and safety of 595-nm pulsed dye laser for the treatment of early superficial infantile hemangioma. Dermatol Ther 35 (5): e15406, 2022.
- Keller RG, Patel KG: Evidence-Based Medicine in the Treatment of Infantile Hemangiomas. Facial Plast Surg Clin North Am 23 (3): 373-92, 2015.
- Sharifpanah F, Saliu F, Bekhite MM, et al.: β-Adrenergic receptor antagonists inhibit vasculogenesis of embryonic stem cells by downregulation of nitric oxide generation and interference with VEGF signalling. Cell Tissue Res 358 (2): 443-52, 2014.
- Ma X, Zhao T, Ouyang T, et al.: Propranolol enhanced adipogenesis instead of induction of apoptosis of hemangiomas stem cells. Int J Clin Exp Pathol 7 (7): 3809-17, 2014.
- Sasaki M, North PE, Elsey J, et al.: Propranolol exhibits activity against hemangiomas independent of beta blockade. NPJ Precis Oncol 3: 27, 2019.
- Overman J, Fontaine F, Wylie-Sears J, et al.: R-propranolol is a small molecule inhibitor of the SOX18 transcription factor in a rare vascular syndrome and hemangioma. Elife 8: , 2019.
- Seebauer CT, Graus MS, Huang L, et al.: Non-beta blocker enantiomers of propranolol and atenolol inhibit vasculogenesis in infantile hemangioma. J Clin Invest 132 (3): , 2022.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al.: A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med 372 (8): 735-46, 2015.
- Bauman NM: Propanolol effectively treats significant infantile hemangiomas. J Pediatr 167 (1): 210, 2015.
- Chang L, Ye X, Qiu Y, et al.: Is Propranolol Safe and Effective for Outpatient Use for Infantile Hemangioma? A Prospective Study of 679 Cases From One Center in China. Ann Plast Surg 76 (5): 559-63, 2016.
- Ames JA, Sykes JM: Current trends in medical management of infantile hemangioma. Curr Opin Otolaryngol Head Neck Surg 23 (4): 286-91, 2015.
- Lou Y, Peng WJ, Cao Y, et al.: The effectiveness of propranolol in treating infantile haemangiomas: a meta-analysis including 35 studies. Br J Clin Pharmacol 78 (1): 44-57, 2014.
- Luo Y, Zeng Y, Zhou B, et al.: A retrospective study of propranolol therapy in 635 infants with infantile hemangioma. Pediatr Dermatol 32 (1): 151-2, 2015 Jan-Feb.
- Vivas-Colmenares GV, Bernabeu-Wittel J, Alonso-Arroyo V, et al.: Effectiveness of propranolol in the treatment of infantile hemangioma beyond the proliferation phase. Pediatr Dermatol 32 (3): 348-52, 2015 May-Jun.
- Kridin K, Pam N, Bergman R, et al.: Oral propranolol administration is effective for infantile hemangioma in late infancy: A retrospective cohort study. Dermatol Ther 33 (3): e13331, 2020.
- Liu X, Qu X, Zheng J, et al.: Effectiveness and Safety of Oral Propranolol versus Other Treatments for Infantile Hemangiomas: A Meta-Analysis. PLoS One 10 (9): e0138100, 2015.
- Hardison S, Wan W, Dodson KM: The use of propranolol in the treatment of subglottic hemangiomas: A literature review and meta-analysis. Int J Pediatr Otorhinolaryngol 90: 175-180, 2016.
- Mehta A, Bajaj MS, Pushker N, et al.: To compare intralesional and oral propranolol for treating periorbital and eyelid capillary hemangiomas. Indian J Ophthalmol 67 (12): 1974-1980, 2019.
- Drolet BA, Frommelt PC, Chamlin SL, et al.: Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics 131 (1): 128-40, 2013.
- Solman L, Glover M, Beattie PE, et al.: Oral propranolol in the treatment of proliferating infantile haemangiomas: British Society for Paediatric Dermatology consensus guidelines. Br J Dermatol 179 (3): 582-589, 2018.
- Hoeger PH, Harper JI, Baselga E, et al.: Treatment of infantile haemangiomas: recommendations of a European expert group. Eur J Pediatr 174 (7): 855-65, 2015.
- Raphael MF, Breugem CC, Vlasveld FA, et al.: Is cardiovascular evaluation necessary prior to and during beta-blocker therapy for infantile hemangiomas?: A cohort study. J Am Acad Dermatol 72 (3): 465-72, 2015.
- Streicher JL, Riley EB, Castelo-Soccio LA: Reevaluating the Need for Electrocardiograms Prior to Initiation of Treatment With Propranolol for Infantile Hemangiomas. JAMA Pediatr 170 (9): 906-7, 2016.
- Püttgen KB, Hansen LM, Lauren C, et al.: Limited utility of repeated vital sign monitoring during initiation of oral propranolol for complicated infantile hemangioma. J Am Acad Dermatol 85 (2): 345-352, 2021.
- Olsen GM, Hansen LM, Stefanko NS, et al.: Evaluating the Safety of Oral Propranolol Therapy in Patients With PHACE Syndrome. JAMA Dermatol 156 (2): 186-190, 2020.
- Prey S, Voisard JJ, Delarue A, et al.: Safety of Propranolol Therapy for Severe Infantile Hemangioma. JAMA 315 (4): 413-5, 2016.
- Morimoto A, Ozeki M, Sasaki S, et al.: Severe hypoglycemia in propranolol treatment for infantile hemangiomas. Pediatr Int 64 (1): e15278, 2022.
- Wedgeworth E, Glover M, Irvine AD, et al.: Propranolol in the treatment of infantile haemangiomas: lessons from the European Propranolol In the Treatment of Complicated Haemangiomas (PITCH) Taskforce survey. Br J Dermatol 174 (3): 594-601, 2016.
- Ji Y, Chen S, Wang Q, et al.: Intolerable side effects during propranolol therapy for infantile hemangioma: frequency, risk factors and management. Sci Rep 8 (1): 4264, 2018.
- Baselga E, Dembowska-Baginska B, Przewratil P, et al.: Efficacy of Propranolol Between 6 and 12 Months of Age in High-Risk Infantile Hemangioma. Pediatrics 142 (3): , 2018.
- Shah SD, Baselga E, McCuaig C, et al.: Rebound Growth of Infantile Hemangiomas After Propranolol Therapy. Pediatrics 137 (4): , 2016.
- Frongia G, Byeon JO, Mehrabi A, et al.: Recurrence rate of infantile hemangioma after oral propranolol therapy. Eur J Pediatr 180 (2): 585-590, 2021.
- O'Brien KF, Shah SD, Pope E, et al.: Late growth of infantile hemangiomas in children >3 years of age: A retrospective study. J Am Acad Dermatol 80 (2): 493-499, 2019.
- Phillips RJ, Crock CM, Penington AJ, et al.: Prolonged tumour growth after treatment of infantile haemangioma with propranolol. Med J Aust 206 (3): 131, 2017.
- Ábarzúa-Araya A, Navarrete-Dechent CP, Heusser F, et al.: Atenolol versus propranolol for the treatment of infantile hemangiomas: a randomized controlled study. J Am Acad Dermatol 70 (6): 1045-9, 2014.
- Bayart CB, Tamburro JE, Vidimos AT, et al.: Atenolol Versus Propranolol for Treatment of Infantile Hemangiomas During the Proliferative Phase: A Retrospective Noninferiority Study. Pediatr Dermatol 34 (4): 413-421, 2017.
- Pope E, Lara-Corrales I, Sibbald C, et al.: Noninferiority and Safety of Nadolol vs Propranolol in Infants With Infantile Hemangioma: A Randomized Clinical Trial. JAMA Pediatr 176 (1): 34-41, 2022.
- Ji Y, Wang Q, Chen S, et al.: Oral atenolol therapy for proliferating infantile hemangioma: A prospective study. Medicine (Baltimore) 95 (24): e3908, 2016.
- McGillis E, Baumann T, LeRoy J: Death Associated With Nadolol for Infantile Hemangioma: A Case for Improving Safety. Pediatrics 145 (1): , 2020.
- Bernabeu-Wittel J, Narváez-Moreno B, de la Torre-García JM, et al.: Oral Nadolol for Children with Infantile Hemangiomas and Sleep Disturbances with Oral Propranolol. Pediatr Dermatol 32 (6): 853-7, 2015 Nov-Dec.
- Chinnadurai S, Fonnesbeck C, Snyder KM, et al.: Pharmacologic Interventions for Infantile Hemangioma: A Meta-analysis. Pediatrics 137 (2): e20153896, 2016.
- Xu DP, Cao RY, Tong S, et al.: Topical timolol maleate for superficial infantile hemangiomas: an observational study. J Oral Maxillofac Surg 73 (6): 1089-94, 2015.
- Tawfik AA, Alsharnoubi J: Topical timolol solution versus laser in treatment of infantile hemangioma: a comparative study. Pediatr Dermatol 32 (3): 369-76, 2015 May-Jun.
- Püttgen K, Lucky A, Adams D, et al.: Topical Timolol Maleate Treatment of Infantile Hemangiomas. Pediatrics 138 (3): , 2016.
- Drolet BA, Boakye-Agyeman F, Harper B, et al.: Systemic timolol exposure following topical application to infantile hemangiomas. J Am Acad Dermatol 82 (3): 733-736, 2020.
- Weibel L, Barysch MJ, Scheer HS, et al.: Topical Timolol for Infantile Hemangiomas: Evidence for Efficacy and Degree of Systemic Absorption. Pediatr Dermatol 33 (2): 184-90, 2016 Mar-Apr.
- Frommelt P, Juern A, Siegel D, et al.: Adverse Events in Young and Preterm Infants Receiving Topical Timolol for Infantile Hemangioma. Pediatr Dermatol 33 (4): 405-14, 2016.
- Muñoz-Garza FZ, Ríos M, Roé-Crespo E, et al.: Efficacy and Safety of Topical Timolol for the Treatment of Infantile Hemangioma in the Early Proliferative Stage: A Randomized Clinical Trial. JAMA Dermatol 157 (5): 583-587, 2021.
- Aly MM, Hamza AF, Abdel Kader HM, et al.: Therapeutic superiority of combined propranolol with short steroids course over propranolol monotherapy in infantile hemangioma. Eur J Pediatr 174 (11): 1503-9, 2015.
- Li G, Xu DP, Tong S, et al.: Oral Propranolol With Topical Timolol Maleate Therapy for Mixed Infantile Hemangiomas in Oral and Maxillofacial Regions. J Craniofac Surg 27 (1): 56-60, 2016.
- Tong S, Xu DP, Liu ZM, et al.: Evaluation of the efficacy and safety of topical timolol maleate combined with oral propranolol treatment for parotid mixed infantile hemangiomas. Oncol Lett 12 (3): 1806-1810, 2016.
- Ge J, Zheng J, Zhang L, et al.: Oral propranolol combined with topical timolol for compound infantile hemangiomas: a retrospective study. Sci Rep 6: 19765, 2016.
- Frieden IJ, Püttgen KB, Drolet BA, et al.: Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol 37 (3): 412-418, 2020.
- Maguiness S, Uihlein LC, Liang MG, et al.: Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol 32 (3): 321-6, 2015 May-Jun.
- Scalise R, Bolton J, Gibbs NF: Rapidly involuting congenital hemangioma (RICH): a brief case report. Dermatol Online J 20 (11): , 2014.
- Kumarasamy MT, Castrisios G, Sharma BK: Rapidly involuting congenital haemangioma in a term neonate. BMJ Case Rep 2014: , 2014.
- Hughes R, McAleer M, Watson R, et al.: Rapidly involuting congenital hemangioma with pustules: two cases. Pediatr Dermatol 31 (3): 398-400, 2014 May-Jun.
- Waelti SL, Rypens F, Damphousse A, et al.: Ultrasound findings in rapidly involuting congenital hemangioma (RICH) - beware of venous ectasia and venous lakes. Pediatr Radiol 48 (4): 586-593, 2018.
- Nasseri E, Piram M, McCuaig CC, et al.: Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol 70 (1): 75-9, 2014.
- Lee PW, Frieden IJ, Streicher JL, et al.: Characteristics of noninvoluting congenital hemangioma: a retrospective review. J Am Acad Dermatol 70 (5): 899-903, 2014.
- Enjolras O, Mulliken JB, Boon LM, et al.: Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg 107 (7): 1647-54, 2001.
- Vildy S, Macher J, Abasq-Thomas C, et al.: Life-threatening hemorrhaging in neonatal ulcerated congenital hemangioma: two case reports. JAMA Dermatol 151 (4): 422-5, 2015.
- Ayturk UM, Couto JA, Hann S, et al.: Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am J Hum Genet 98 (4): 789-95, 2016.
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- International Society for the Study of Vascular Anomalies: ISSVA Classification of Vascular Anomalies. Milwaukee, Wi: International Society for the Study of Vascular Anomalies, 2018. Available online. Last accessed June 7, 2022.
- Wassef M, Blei F, Adams D, et al.: Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 136 (1): e203-14, 2015.
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971.
- Kulungowski AM, Alomari AI, Chawla A, et al.: Lessons from a liver hemangioma registry: subtype classification. J Pediatr Surg 47 (1): 165-70, 2012.
- Berklite L, Malik F, Ranganathan S, et al.: Pediatric hepatic vascular tumors: clinicopathologic characteristics of 33 cases and proposed updates to current classification schemes. Hum Pathol 141: 78-89, 2023.
- Kayaalp C, Sabuncuoglu MZ: Embolization of Liver Hemangiomas. Hepat Mon 15 (8): e30334, 2015.
- Klein M, Chang AK, Vasudevan SA, et al.: Clinically significant ascites as an indication for resection of rapidly involuting congenital hepatic hemangiomas. Pediatr Blood Cancer 65 (8): e27222, 2018.
- Gourgiotis S, Moustafellos P, Zavos A, et al.: Surgical treatment of hepatic haemangiomas: a 15-year experience. ANZ J Surg 76 (9): 792-5, 2006.
- Schmitz R, Heinig J, Klockenbusch W, et al.: Antenatal diagnosis of a giant fetal hepatic hemangioma and treatment with maternal corticosteroid. Ultraschall Med 30 (3): 223-6, 2009.
- Rialon KL, Murillo R, Fevurly RD, et al.: Impact of Screening for Hepatic Hemangiomas in Patients with Multiple Cutaneous Infantile Hemangiomas. Pediatr Dermatol 32 (6): 808-12, 2015 Nov-Dec.
- Yeh I, Bruckner AL, Sanchez R, et al.: Diffuse infantile hepatic hemangiomas: a report of four cases successfully managed with medical therapy. Pediatr Dermatol 28 (3): 267-75, 2011 May-Jun.
- Wasserman JD, Mahant S, Carcao M, et al.: Vincristine for successful treatment of steroid-dependent infantile hemangiomas. Pediatrics 135 (6): e1501-5, 2015.
- Vlahovic A, Simic R, Djokic D, et al.: Diffuse neonatal hemangiomatosis treatment with cyclophosphamide: a case report. J Pediatr Hematol Oncol 31 (11): 858-60, 2009.
- Sundar Alagusundaramoorthy S, Vilchez V, Zanni A, et al.: Role of transplantation in the treatment of benign solid tumors of the liver: a review of the United Network of Organ Sharing data set. JAMA Surg 150 (4): 337-42, 2015.
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014.
- Rutten C, Ackermann O, Lambert V, et al.: Pediatric hepatic hemangiomas: spectrum and prognostic significance of initial ultrasound findings. Pediatr Radiol 53 (12): 2446-2457, 2023.
- Sari N, Yalçin B, Akyüz C, et al.: Infantile hepatic hemangioendothelioma with elevated serum alpha-fetoprotein. Pediatr Hematol Oncol 23 (8): 639-47, 2006.
- Seo IS, Min KW, Mirkin LD: Hepatic hemangioendothelioma of infancy associated with elevated alpha fetoprotein and catecholamine by-products. Pediatr Pathol 8 (6): 625-31, 1988.
- Langham MR, Furman WL, Fernandez-Pineda I: Current Management of Neonatal Liver Tumors. Curr Pediatr Rev 11 (3): 195-204, 2015.
- Perkins P, Weiss SW: Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol 20 (10): 1196-204, 1996.
- Fletcher CD, Beham A, Schmid C: Spindle cell haemangioendothelioma: a clinicopathological and immunohistochemical study indicative of a non-neoplastic lesion. Histopathology 18 (4): 291-301, 1991.
- Enjolras O, Mulliken JB, Kozakewich HPW: Vascular tumors and tumor-like lesions. In: Mulliken JB, Burrows PE, Fishman SJ, eds.: Mulliken & Young's Vascular Anomalies: Hemangiomas and Malformations. 2nd ed. Oxford University Press, 2013, pp 259-324.
- Hoeger PH, Colmenero I: Vascular tumours in infants. Part I: benign vascular tumours other than infantile haemangioma. Br J Dermatol 171 (3): 466-73, 2014.
- Pansuriya TC, van Eijk R, d'Adamo P, et al.: Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet 43 (12): 1256-61, 2011.
- Guo R, Gavino AC: Angiolymphoid hyperplasia with eosinophilia. Arch Pathol Lab Med 139 (5): 683-6, 2015.
- O'Connell JX, Nielsen GP, Rosenberg AE: Epithelioid vascular tumors of bone: a review and proposal of a classification scheme. Adv Anat Pathol 8 (2): 74-82, 2001.
- Huang SC, Zhang L, Sung YS, et al.: Frequent FOS Gene Rearrangements in Epithelioid Hemangioma: A Molecular Study of 58 Cases With Morphologic Reappraisal. Am J Surg Pathol 39 (10): 1313-21, 2015.
- Liu KX, Duggan EM, Al-Ibraheemi A, et al.: Characterization of long-term outcomes for pediatric patients with epithelioid hemangioma. Pediatr Blood Cancer 66 (1): e27451, 2019.
- Browning JC, Eldin KW, Kozakewich HP, et al.: Congenital disseminated pyogenic granuloma. Pediatr Dermatol 26 (3): 323-7, 2009 May-Jun.
- Putra J, Rymeski B, Merrow AC, et al.: Four cases of pediatric deep-seated/subcutaneous pyogenic granuloma: Review of literature and differential diagnosis. J Cutan Pathol 44 (6): 516-522, 2017.
- Alomari MH, Kozakewich HPW, Kerr CL, et al.: Congenital Disseminated Pyogenic Granuloma: Characterization of an Aggressive Multisystemic Disorder. J Pediatr 226: 157-166, 2020.
- Groesser L, Peterhof E, Evert M, et al.: BRAF and RAS Mutations in Sporadic and Secondary Pyogenic Granuloma. J Invest Dermatol 136 (2): 481-6, 2016.
- Lee J, Sinno H, Tahiri Y, et al.: Treatment options for cutaneous pyogenic granulomas: a review. J Plast Reconstr Aesthet Surg 64 (9): 1216-20, 2011.
- Patrizi A, Gurioli C, Dika E: Pyogenic granulomas in childhood: New treatment modalities. Dermatol Ther 28 (5): 332, 2015 Sep-Oct.
- Oke I, Alkharashi M, Petersen RA, et al.: Treatment of Ocular Pyogenic Granuloma With Topical Timolol. JAMA Ophthalmol 135 (4): 383-385, 2017.
- Jaiswal H, Patidar N, Shah C, et al.: Topical timolol 0.5% as the primary treatment of ophthalmic pyogenic granuloma: A prospective, single-arm study. Indian J Ophthalmol 69 (5): 1155-1160, 2021.
- Neri I, Baraldi C, Balestri R, et al.: Topical 1% propranolol ointment with occlusion in treatment of pyogenic granulomas: An open-label study in 22 children. Pediatr Dermatol 35 (1): 117-120, 2018.
- Haemel AK, O'Brian AL, Teng JM: Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol 146 (7): 715-8, 2010.
- Pignatti M, Spaggiari A, Sala P, et al.: Laser treatment of angiofibromas in tuberous sclerosis. Minerva Pediatr 66 (6): 585-6, 2014.
- Lee YI, Lee JH, Kim DY, et al.: Comparative Effects of Topical 0.2% Sirolimus for Angiofibromas in Adults and Pediatric Patients with Tuberous Sclerosis Complex. Dermatology 234 (1-2): 13-22, 2018.
- Wataya-Kaneda M, Ohno Y, Fujita Y, et al.: Sirolimus Gel Treatment vs Placebo for Facial Angiofibromas in Patients With Tuberous Sclerosis Complex: A Randomized Clinical Trial. JAMA Dermatol 154 (7): 781-788, 2018.
- Koenig MK, Bell CS, Hebert AA, et al.: Efficacy and Safety of Topical Rapamycin in Patients With Facial Angiofibromas Secondary to Tuberous Sclerosis Complex: The TREATMENT Randomized Clinical Trial. JAMA Dermatol 154 (7): 773-780, 2018.
- Coutinho-Camillo CM, Brentani MM, Nagai MA: Genetic alterations in juvenile nasopharyngeal angiofibromas. Head Neck 30 (3): 390-400, 2008.
- Riggs S, Orlandi RR: Juvenile nasopharyngeal angiofibroma recurrence associated with exogenous testosterone therapy. Head Neck 32 (6): 812-5, 2010.
- Liu Z, Wang J, Wang H, et al.: Hormonal receptors and vascular endothelial growth factor in juvenile nasopharyngeal angiofibroma: immunohistochemical and tissue microarray analysis. Acta Otolaryngol 135 (1): 51-7, 2015.
- Sakthivel P, Kumar R, Arunraj ST, et al.: 68 Ga DOTANOC PET/CT Scan in Primary Juvenile Nasopharyngeal Angiofibroma - A Pilot Study. Laryngoscope 131 (7): 1509-1515, 2021.
- Szymańska A, Szymański M, Czekajska-Chehab E, et al.: Two types of lateral extension in juvenile nasopharyngeal angiofibroma: diagnostic and therapeutic management. Eur Arch Otorhinolaryngol 272 (1): 159-66, 2015.
- Samanta D: Topical mTOR (mechanistic target of rapamycin) inhibitor therapy in facial angiofibroma. Indian J Dermatol Venereol Leprol 81 (5): 540-1, 2015 Sep-Oct.
- Krakowski AC, Nguyen TA: Inhibition of Angiofibromas in a Tuberous Sclerosis Patient Using Topical Timolol 0.5% Gel. Pediatrics 136 (3): e709-13, 2015.
- Mallick S, Benson R, Bhasker S, et al.: Long-term treatment outcomes of juvenile nasopharyngeal angiofibroma treated with radiotherapy. Acta Otorhinolaryngol Ital 35 (2): 75-9, 2015.
- Peters T, Traboulsi D, Tibbles LA, et al.: Sirolimus: a therapeutic advance for dermatologic disease. Skin Therapy Lett 19 (4): 1-4, 2014 Jul-Aug.
- Fernández KS, de Alarcon A, Adams DM, et al.: Sirolimus for the treatment of juvenile nasopharyngeal angiofibroma. Pediatr Blood Cancer 67 (4): e28162, 2020.
Tumores intermedios (localmente invasores)
Hemangioendotelioma kaposiforme y angioma en penacho
El hemangioendotelioma kaposiforme y el angioma en penacho son tumores vasculares infrecuentes que se presentan de manera típica durante la lactancia o la niñez temprana, pero también se notificaron casos en adultos. Se cree que ambos tumores constituyen variantes de la misma enfermedad, porque ambos pueden ser localmente invasores y causar el fenómeno de Kasabach-Merritt, una coagulopatía grave potencialmente mortal caracterizada por trombocitopenia profunda e hipofibrinogenemia. En este texto se tratan como una sola entidad: hemangioendotelioma kaposiforme.
Incidencia
Aunque no se conoce la incidencia exacta del hemangioendotelioma kaposiforme, se estima que sea de 0,07 casos anuales por cada 100 000 niños.[1,2,3] Esta lesión afecta ambos sexos por igual; la mayoría surge en el período neonatal, la mitad está presente en el nacimiento y otras se manifiestan durante la infancia o la edad adulta.[4]
Cuadro clínico inicial
El hemangioendotelioma kaposiforme por lo general compromete las extremidades; es menos frecuente que afecte el tronco y el área de la cabeza y el cuello.[3] La mayoría de las lesiones afectan la piel (consultar la Figura 8). Es posible que las lesiones más profundas (retroperitoneo, cavidad torácica y músculo) se vean de tonalidad azulada-purpúrica sobre la piel, mientras que las lesiones superficiales pueden ser firmes, purpúricas o equimóticas, y dolorosas. Las lesiones óseas primarias tal vez produzcan dolor u otros hallazgos inespecíficos, aunque no haya masa evidente durante el examen físico.[5][Nivel de evidencia C2] Las lesiones suelen ser unifocales y de crecimiento expansivo y contiguo. A veces los ganglios linfáticos locales están comprometidos, pero no se informan metástasis a distancia. Se notificaron presentaciones multifocales infrecuentes, sobre todo en el hueso.[1,2,3]
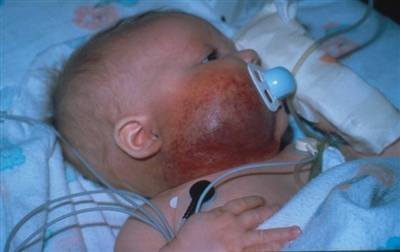
Entre los pacientes con hemangioendotelioma kaposiforme, del 50 % al 70 % presentan el fenómeno de Kasabach-Merritt, una complicación potencialmente mortal. El riesgo de presentar el fenómeno de Kasabach-Merritt es más alto en los pacientes con lesiones congénitas, lesiones de más de 8 cm y cuando el hemangioendotelioma kaposiforme se origina en el retroperitoneo o el mediastino.[3,6] Esta afección se caracteriza por trombocitopenia grave (intervalo, 3000–60 000/µl) e hipofibrinogenemia (<1 g/l). El dímero D y los productos de la degradación de la fibrina están elevados. Se puede presentar anemia grave debido al secuestro tumoral. La hemorragia grave es infrecuente; sin embargo, es posible que un trauma (biopsia, procedimiento quirúrgico), una ulceración, una infección o la demora para iniciar el tratamiento desencadene una coagulación intravascular diseminada, una hemorragia grave y hasta la muerte. El remplazo intensivo de productos sanguíneos, en particular plaquetas, quizás aumente el tamaño de la lesión y cause dolor intenso; solo se deberá considerar ante un sangrado activo y bajo la dirección de un especialista en anomalías vasculares.[3] No está clara la tasa de mortalidad, pero se ha notificado que alcanza hasta un 30 %.[3,6]
Características histopatológicas
El hemangioendotelioma kaposiforme se caracteriza por capas de células fusiformes con un modelo infiltrante en la dermis, la grasa subcutánea y el músculo. A menudo hay áreas de fibrosis, con vasos dilatados de pared delgada infiltrados en torno a las áreas de células fusiformes. Mezcladas dentro de estas áreas hay focos de células epitelioides redondeadas de origen vascular y conjuntos de capilares con lúmenes de forma redondeada o irregular que contienen trombos ricos en plaquetas y fibrina. Por lo general, hay espacios linfáticos anormales dentro de la lesión o en la periferia. La tasa de mitosis es variable, pero suele ser baja. El angioma en penacho se caracteriza por múltiples lobulillos independientes formados por grupos densos de capilares (penachos) dispersos en la dermis y, a veces, en la hipodermis, en una configuración conocida como bala de cañón.[7] Las mitosis son poco frecuentes.
Se tiene poco conocimiento sobre la patogenia. Hay algunos indicios de que el hemangioendotelioma kaposiforme se deriva del endotelio linfático, porque las células fusiformes expresan los marcadores vasculares CD31 y CD34, el receptor del factor de crecimiento endotelial vascular-3 (VEGFR-3) (receptor necesario para la linfangiogénesis) y los marcadores linfáticos D2-40 y PROX1.[7,8,9] No hay evidencia de una relación con la infección por el virus del herpes humano 8 como en el sarcoma de Kaposi.[9]
Los datos genómicos son escasos. Se notificó una pequeña cantidad de pacientes con variantes de GNA14, pero no en todos los casos.[10,11]
Se han encontrado concentraciones séricas altas de angiopoyetina-2 (Ang-2) en pacientes de riesgo alto con hemangioendotelioma kaposiforme y linfangiomatosis kaposiforme. También se observó que las concentraciones de Ang-2 disminuyeron como respuesta al tratamiento con sirólimus, lo que aumenta la posibilidad de que tenga un efecto en las células endoteliales del hemangioendotelioma kaposiforme.[12] Las células endoteliales producen y almacenan Ang-2, que actúa como un antagonista de tirosina–cinasa TEK. Es posible que Ang-2 favorezca la neovascularización junto con el VEGF. En los seres humanos, la Ang-2 aumenta mucho durante la remodelación vascular que ocurre con la sepsis, la inflamación y la linfangiomatosis.[13] Estas concentraciones se han utilizado para diagnosticar tumores vasculares y para evaluar la respuesta al tratamiento.
Evaluación diagnóstica
Para el diagnóstico se tiene en cuenta la combinación de las características clínicas, histológicas y de imágenes. La evaluación de laboratorio es esencial para el diagnóstico del fenómeno de Kasabach-Merritt. Siempre que sea posible, se debe obtener confirmación histológica porque a menudo se necesita un tratamiento prolongado. Sin embargo, si los hallazgos clínicos y de imágenes son muy indicativos del diagnóstico, aplazar la biopsia quizás sea una opción, pero esto debe decidirse mediante un análisis y enfoque interdisciplinarios.
Las imágenes por resonancia magnética (IRM) son la modalidad de imagen preferida, en especial, para el hemangioendotelioma kaposiforme que se presenta con el fenómeno de Kasabach-Merritt y lesiones grandes. Las secuencias ponderadas en T1 por lo general muestran una masa de tejido blando mal circunscrito con engrosamiento del tejido blando y dérmico, y realce difuso con gadolinio. En las secuencias ponderadas en T2, se observa un aumento difuso en la señal, con trabeculación en la grasa subcutánea. Las secuencias de gradiente muestran los vasos ligeramente dilatados en la masa de tejido blando y alrededor de esta.[3]
En las lesiones pequeñas y superficiales, es posible que la ecografía sea útil para el diagnóstico, además, esta ayuda a diferenciar el angioma en penacho del hemangioendotelioma kaposiforme. Los angiomas en penacho son hiperecoicos, más superficiales y tienen bordes bien definidos. El hemangioendotelioma kaposiforme tiene una configuración más infiltrante , con bordes mal definidos y ecogenicidad mixta. También presentan más densidad vascular que los angiomas en penacho.[14]
Tratamiento del hemangioendotelioma kaposiforme y el angioma en penacho
Tratamiento del hemangioendotelioma kaposiforme y el angioma en penacho sin complicaciones
No hay un estándar de atención que se fundamente en la evidencia científica para los hemangioendoteliomas kaposiformes ni para los angiomas en penacho. El tratamiento varía según el tamaño, la localización, la presencia de síntomas y la gravedad de la coagulopatía.
Las opciones de tratamiento para los hemangioendoteliomas kaposiformes y los angiomas en penacho sin complicaciones son las siguientes:
- Observación.
- Escisión quirúrgica.
- Láser de colorante pulsado.
- Fármacos tópicos.
- Propranolol.
- Sirólimus con terapia de corticoesteroides o sin esta.
La observación es una opción para los pacientes con tumores de riesgo bajo (es decir, sin el fenómeno de Kasabach-Merritt, tumores pequeños, asintomáticos). Se ha notificado regresión espontánea o estabilidad.[15]
Es posible tratar con escisión quirúrgica, láser de colorante pulsado o fármacos tópicos (esteroides, sirólimus o tacrólimus) los hemangioendoteliomas kaposiformes y los angiomas en penacho localizados que no son complicados.[15,16,17]
Se notificó que el propranolol es una opción de tratamiento para los pacientes con hemangioendoteliomas kaposiformes según los resultados positivos que se obtuvieron con el uso de este medicamento en otros tumores vasculares más benignos. Los resultados han sido contradictorios; un informe señala mejora de la eficacia con dosis más altas de propranolol.[18,19] En los resultados preliminares se indica que el propranolol se debe reservar para los pacientes con hemangioendoteliomas kaposiformes sin el fenómeno de Kasabach-Merritt y con lesiones más pequeñas, menos complicadas.
Tratamiento del hemangioendotelioma kaposiforme y el angioma en penacho complicados
Los pacientes con el fenómeno de Kasabach-Merritt o compromiso funcional y que presentan síntomas necesitan tratamiento intensivo. Un panel multidisciplinario de expertos de los Estados Unidos y Canadá publicó directrices para el tratamiento de los hemangioendoteliomas kaposiformes complicados.[20] Se notificaron varias opciones de tratamiento, pero ninguna ha sido eficaz en todos los casos.[21,22]
Las opciones de tratamiento de los hemangioendoteliomas kaposiformes complicados y con el fenómeno de Kasabach-Merritt son las siguientes:
- Vincristina con terapia con corticoesteroides o sin esta.
- Sirólimus con terapia de corticoesteroides o sin esta.
- Escisión quirúrgica.
Vincristina con terapia con corticoesteroides o sin esta
El tratamiento más común para los hemangioendoteliomas kaposiformes complicados con el fenómeno de Kasabach-Merritt o sin este ha sido, tradicionalmente, la terapia con corticoesteroides con vincristina u otros fármacos o sin estos.[20,21,22,23,24,25] Sin embargo, muchas instituciones ahora usan el inhibidor de mTOR, sirólimus, con terapia con corticoesteroides o sin esta, como tratamiento primario para los pacientes de riesgo alto.[26,27,28,29,30] La terapia con corticoesteroides no ha mostrado ser eficaz como monoterapia para los hemangioendoteliomas kaposiformes complicados, así se use en dosis altas. Los pacientes que reciben terapia con corticoesteroides tienen una tasa de respuesta del 10 % al 20 % y una cantidad importante de efectos secundarios.[20]
Se observó una respuesta hematológica y una reducción del volumen tumoral en pacientes con hemangioendoteliomas kaposiformes de riesgo alto con el uso de vincristina.[21] Además, en una revisión retrospectiva de 37 niños con hemangioendoteliomas kaposiformes en cuyas lesiones no se observó respuesta a los esteroides, se notificó remisión completa en 26 lesiones, con recuentos plaquetarios que alcanzaron las concentraciones normales dentro de las 7,6 (± 5.2) semanas posteriores al tratamiento con vincristina.[23][Nivel de evidencia C3] En otros estudios, la vincristina en monoterapia no ha mostrado ser eficaz.[26,30] También se ha notificado el tratamiento exitoso de pacientes con hemangioendoteliomas kaposiformes que recibieron vincristina y ticlopidina.[31]
En 2013, se propuso en las pautas consensuadas para el tratamiento de los hemangioendoteliomas kaposiformes complicados, sobre la base de la evidencia disponible, el uso de vincristina con esteroides o sin estos como terapia de primera línea. Esta recomendación se fundamentó en la evidencia disponible.[20]
Sirólimus con terapia de corticoesteroides o sin esta
A partir de informes de casos, series de casos y un ensayo clínico prospectivo promisorios, es posible que se considere el sirólimus como una terapia de primera línea alternativa para los pacientes con hemangioendoteliomas kaposiformes.[27,28,32] Hay pocos estudios en los que se investigue el efecto del sirólimus en los hemangioendoteliomas kaposiformes o los angiomas en penacho sin el fenómeno de Kasabach-Merritt.
Evidencia (tratamiento con sirólimus):
- En un estudio prospectivo en el que se evalúo la eficacia e inocuidad del sirólimus para el tratamiento de las anomalías vasculares complicadas, 13 pacientes con hemangioendoteliomas kaposiformes recibieron sirólimus.[30]
- Entre los pacientes con hemangioendoteliomas kaposiformes y el fenómeno de Kasabach-Merritt, 10 de 10 pacientes presentaron respuestas parciales con normalización del recuento de plaquetas y el fibrinógeno al final de 6 y 12 cursos.
- De los 3 pacientes con hemangioendoteliomas kaposiformes sin el fenómeno de Kasabach-Merritt, 2 pacientes presentaron respuestas parciales al final del curso 12. El l tercer paciente con enfermedad multifocal ósea presentó progresión de la enfermedad.
- Los efectos secundarios fueron mínimos en este grupo de pacientes jóvenes y ningún paciente con un hemangioendotelioma kaposiforme necesitó ajuste de la dosis o se retiró del estudio debido a efectos tóxicos del sirólimus.
- En un estudio retrospectivo del tratamiento con sirólimus en pacientes cuya mayoría habían recibido otros tratamientos antes, se notificó una tasa de respuesta completa del 73 %. En todos los pacientes cuya evaluación indicó la presencia del fenómeno de Kasabach-Merritt, se presentó una mejora de los recuentos plaquetarios entre el día 1 y la semana 3 (media, 1,3 semanas).[33]
- En este estudio, hubo 1 muerte por infección respiratoria en un niño con efusión pleural y enfermedad multifocal que comprometió la cavidad torácica. No se hicieron biopsias en forma rutinaria para confirmar diagnósticos, lo que aumenta la posibilidad de que este niño presentara una anomalía linfática compleja desconocida.
- En un estudio de cohortes retrospectivo multicéntrico, se examinaron a 52 pacientes chinos con hemangioendoteliomas kaposiformes progresivos. Entre estos, 37 pacientes (71 %) tenían el fenómeno de Kasabach-Merritt. Aquellos sin el fenómeno de Kasabach-Merritt recibieron sirólimus solo y 21 pacientes con el fenómeno de Kasabach-Merritt recibieron una combinación de sirólimus y prednisona.[29]
- En general, el 96 % de los pacientes a los 6 meses y el 98 % de los pacientes a los 12 meses presentaron una mejora de los síntomas observables o una mejora de las complicaciones.
- En un solo informe de caso, se describió un niño con hemangioendotelioma kaposiforme que presentó recidiva del dolor y fibrosis años después de la terapia inicial y que se trató con sirólimus durante 26 meses; se observaron los siguientes resultados:[32]
- La contractura y la amplitud de movimiento del paciente mejoraron, la lesión se achicó y el niño estaba bien 2 años después.
- En un estudio prospectivo aleatorizado de pacientes con hemangioendotelioma kaposiforme relacionado con el fenómeno de Kasabach-Merritt, se comparó el sirólimus en monoterapia con el sirólimus combinado con prednisolona. El resultado primario se definió como el logro de una respuesta plaquetaria duradera (recuento de plaquetas >100 × 109 /l) en la semana 4.[34][Nivel de evidencia A3]
- En la semana 4, se logró una respuesta plaquetaria duradera en 35 de 37 pacientes que recibieron sirólimus y prednisolona, en comparación con 24 de 36 pacientes que recibieron sirólimus en monoterapia (diferencia, 27,9 %; IC 95 %, 10,0–44,7 %).
- En comparación con el grupo de monoterapia con sirólimus, el grupo de tratamiento combinado mostró mejoras en las mediciones de respuestas plaquetarias duraderas en todos los puntos durante el periodo de tratamiento inicial de 3 semanas. Las respuestas plaquetarias duraderas incluyeron mediana de recuento de plaquetas durante las semanas 1 a 4, un mayor número de pacientes que lograron la estabilización del fibrinógeno en la semana 4 y respuestas objetivas de las lesiones a los 12 meses.
- Las frecuencias de eventos adversos graves y totales fueron similares en ambos grupos.
- Los pacientes que recibieron el tratamiento combinado tuvieron menos secuelas totales de enfermedad y menos transfusiones sanguíneas.
La mayoría de pacientes de riesgo alto (hemangioendotelioma kaposiforme con fenómeno de Kasabach-Merritt), se tratan con sirólimus para lograr concentraciones séricas de 8 ng/ml a 15 ng/ml.[29,30,35,36]
Cuidados médicos de apoyo y seguimiento minucioso de los lactantes que reciben sirólimus
En un informe de caso, se describió a 2 niños con hemangioendoteliomas kaposiformes y síndrome de Kasabach-Merritt que murieron por infecciones pulmonares después del tratamiento con sirólimus.[37] Otro niño que recibió sirólimus y prednisolona presentó neumonía por Pneumocystis jirovecii.[38] Se recomienda la profilaxis para la neumonía por P. jirovecci y el seguimiento minucioso de los pacientes que reciben sirólimus (en especial lactantes).
Escisión quirúrgica
Quizá sea posible la escisión quirúrgica de las lesiones que no responden al tratamiento médico o que ponen en peligro la vida. La embolización se puede realizar junto con la cirugía o la terapia médica; por lo general, se trata de una medida temporal.[39]
Desenlaces a largo plazo
Aún después del tratamiento, estas lesiones no remiten por completo y es posible que recidiven. Los síntomas (dolor e inflamación) empeoran con la edad; en especial, alrededor de la pubertad.[40]
Los efectos a largo plazo son dolor crónico, linfedema, insuficiencia cardíaca y problemas ortopédicos.[39,40] Estas lesiones constituyen un dilema difícil para el médico porque sus manifestaciones clínicas y la respuesta al tratamiento son variadas.
Opciones de tratamiento en evaluación clínica para el hemangioendotelioma kaposiforme
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Referencias:
- Rodriguez V, Lee A, Witman PM, et al.: Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 31 (7): 522-6, 2009.
- Ryan C, Price V, John P, et al.: Kasabach-Merritt phenomenon: a single centre experience. Eur J Haematol 84 (2): 97-104, 2010.
- Croteau SE, Liang MG, Kozakewich HP, et al.: Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr 162 (1): 142-7, 2013.
- Lee B, Chiu M, Soriano T, et al.: Adult-onset tufted angioma: a case report and review of the literature. Cutis 78 (5): 341-5, 2006.
- Kuo C, Warren M, Malvar J, et al.: Kaposiform hemangioendothelioma of the bone in children and adolescents. Pediatr Blood Cancer 69 (1): e29392, 2022.
- Ji Y, Yang K, Peng S, et al.: Kaposiform haemangioendothelioma: clinical features, complications and risk factors for Kasabach-Merritt phenomenon. Br J Dermatol 179 (2): 457-463, 2018.
- Enjolras O, Soupre V, Picard A: Uncommon benign infantile vascular tumors. Adv Dermatol 24: 105-24, 2008.
- Zukerberg LR, Nickoloff BJ, Weiss SW: Kaposiform hemangioendothelioma of infancy and childhood. An aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 17 (4): 321-8, 1993.
- Arai E, Kuramochi A, Tsuchida T, et al.: Usefulness of D2-40 immunohistochemistry for differentiation between kaposiform hemangioendothelioma and tufted angioma. J Cutan Pathol 33 (7): 492-7, 2006.
- Lim YH, Bacchiocchi A, Qiu J, et al.: GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation. Am J Hum Genet 99 (2): 443-50, 2016.
- Lim YH, Fraile C, Antaya RJ, et al.: Tufted angioma with associated Kasabach-Merritt phenomenon caused by somatic mutation in GNA14. Pediatr Dermatol 36 (6): 963-964, 2019.
- Le Cras TD, Mobberley-Schuman PS, Broering M, et al.: Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis 20 (1): 163-173, 2017.
- Saharinen P, Eklund L, Alitalo K: Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 16 (9): 635-661, 2017.
- Gong X, Ying H, Zhang Z, et al.: Ultrasonography and magnetic resonance imaging features of kaposiform hemangioendothelioma and tufted angioma. J Dermatol 46 (10): 835-842, 2019.
- Osio A, Fraitag S, Hadj-Rabia S, et al.: Clinical spectrum of tufted angiomas in childhood: a report of 13 cases and a review of the literature. Arch Dermatol 146 (7): 758-63, 2010.
- Burleigh A, Kanigsberg N, Lam JM: Topical rapamycin (sirolimus) for the treatment of uncomplicated tufted angiomas in two children and review of the literature. Pediatr Dermatol 35 (5): e286-e290, 2018.
- Zhang X, Yang K, Chen S, et al.: Tacrolimus ointment for the treatment of superficial kaposiform hemangioendothelioma and tufted angioma. J Dermatol 46 (10): 898-901, 2019.
- Filippi L, Tamburini A, Berti E, et al.: Successful Propranolol Treatment of a Kaposiform Hemangioendothelioma Apparently Resistant to Propranolol. Pediatr Blood Cancer 63 (7): 1290-2, 2016.
- Wang Z, Li K, Dong K, et al.: Variable response to propranolol treatment of kaposiform hemangioendothelioma, tufted angioma, and Kasabach-Merritt phenomenon. Pediatr Blood Cancer 61 (8): 1518-9, 2014.
- Drolet BA, Trenor CC, Brandão LR, et al.: Consensus-derived practice standards plan for complicated Kaposiform hemangioendothelioma. J Pediatr 163 (1): 285-91, 2013.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al.: Kasabach-merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 24 (6): 459-62, 2002 Aug-Sep.
- Hauer J, Graubner U, Konstantopoulos N, et al.: Effective treatment of kaposiform hemangioendotheliomas associated with Kasabach-Merritt phenomenon using four-drug regimen. Pediatr Blood Cancer 49 (6): 852-4, 2007.
- Wang Z, Li K, Yao W, et al.: Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients treated with vincristine and long-term follow-up. Pediatr Blood Cancer 62 (4): 577-80, 2015.
- Fernandez-Pineda I, Lopez-Gutierrez JC, Ramirez G, et al.: Vincristine-ticlopidine-aspirin: an effective therapy in children with Kasabach-Merritt phenomenon associated with vascular tumors. Pediatr Hematol Oncol 27 (8): 641-5, 2010.
- Fernandez-Pineda I, Lopez-Gutierrez JC, Chocarro G, et al.: Long-term outcome of vincristine-aspirin-ticlopidine (VAT) therapy for vascular tumors associated with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 60 (9): 1478-81, 2013.
- Kai L, Wang Z, Yao W, et al.: Sirolimus, a promising treatment for refractory Kaposiform hemangioendothelioma. J Cancer Res Clin Oncol 140 (3): 471-6, 2014.
- Hammill AM, Wentzel M, Gupta A, et al.: Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer 57 (6): 1018-24, 2011.
- Blatt J, Stavas J, Moats-Staats B, et al.: Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer 55 (7): 1396-8, 2010.
- Ji Y, Chen S, Xiang B, et al.: Sirolimus for the treatment of progressive kaposiform hemangioendothelioma: A multicenter retrospective study. Int J Cancer 141 (4): 848-855, 2017.
- Adams DM, Trenor CC, Hammill AM, et al.: Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics 137 (2): e20153257, 2016.
- López V, Martí N, Pereda C, et al.: Successful management of Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon using vincristine and ticlopidine. Pediatr Dermatol 26 (3): 365-6, 2009 May-Jun.
- Oza VS, Mamlouk MD, Hess CP, et al.: Role of Sirolimus in Advanced Kaposiform Hemangioendothelioma. Pediatr Dermatol 33 (2): e88-92, 2016 Mar-Apr.
- Wang Z, Yao W, Sun H, et al.: Sirolimus therapy for kaposiform hemangioendothelioma with long-term follow-up. J Dermatol 46 (11): 956-961, 2019.
- Ji Y, Chen S, Zhou J, et al.: Sirolimus plus prednisolone vs sirolimus monotherapy for kaposiform hemangioendothelioma: a randomized clinical trial. Blood 139 (11): 1619-1630, 2022.
- Zhang G, Chen H, Gao Y, et al.: Sirolimus for treatment of Kaposiform haemangioendothelioma with Kasabach-Merritt phenomenon: a retrospective cohort study. Br J Dermatol 178 (5): 1213-1214, 2018.
- Mariani LG, Schmitt IR, Garcia CD, et al.: Low dose sirolimus treatment for refractory tufted angioma and congenital kaposiform hemangioendothelioma, both with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 66 (8): e27810, 2019.
- Ying H, Qiao C, Yang X, et al.: A Case Report of 2 Sirolimus-Related Deaths Among Infants With Kaposiform Hemangioendotheliomas. Pediatrics 141 (Suppl 5): S425-S429, 2018.
- Russell TB, Rinker EK, Dillingham CS, et al.: Pneumocystis Jirovecii Pneumonia During Sirolimus Therapy for Kaposiform Hemangioendothelioma. Pediatrics 141 (Suppl 5): S421-S424, 2018.
- Ji Y, Chen S, Yang K, et al.: Kaposiform hemangioendothelioma: current knowledge and future perspectives. Orphanet J Rare Dis 15 (1): 39, 2020.
- Schaefer BA, Wang D, Merrow AC, et al.: Long-term outcome for kaposiform hemangioendothelioma: A report of two cases. Pediatr Blood Cancer 64 (2): 284-286, 2017.

