Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre la leucemia linfocítica crónica
Incidencia y mortalidad
Número estimado de casos nuevos y defunciones por la leucemia linfocítica crónica (LLC) en los Estados Unidos en 2025:[1]
Casos nuevos: 23 690.
Defunciones: 4460.
Características anatómicas
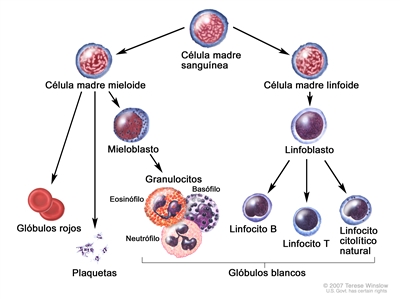
La LLC es un trastorno de linfocitos morfológicamente maduros pero inmunológicamente inmaduros, que se manifiesta por la acumulación progresiva de estas células en la sangre, la médula ósea y los tejidos linfáticos.[2]
Cuadro clínico inicial
La evolución clínica de esta enfermedad progresa desde una linfocitosis de escasa malignidad sin otros indicios de enfermedad hasta una hiperplasia linfática generalizada con pancitopenia simultánea. Las complicaciones de la pancitopenia incluyen hemorragia e infección, que son unas de las causas principales de muerte en estos pacientes.[3] La alteraciones inmunológicas, como la anemia hemolítica con resultado positivo en la prueba de Coombs, la trombocitopenia inmunitaria y las concentraciones bajas de inmunoglobulinas, complican el tratamiento de la LLC.[4]
Evaluación diagnóstica y diagnóstico diferencial
Las pruebas y procedimientos que se usan para el diagnóstico de la LLC son los siguientes:[5]
Antecedentes y examen físico (incluyendo los diámetros bidimensionales de los ganglios linfáticos palpables más grandes en los sitios ganglionares cervicales, axilares e inguinales, y las dimensiones del hígado y el bazo debajo de sus respectivos márgenes costales según se determinó en la palpación).
Recuento sanguíneo completo con diferencial y estudios bioquímicos (incluso creatinina, bilirrubina, transaminasas y fosfatasa alcalina). Otras pruebas sanguíneas que a veces se usan son la concentración de lactato-deshidrogenasa y de β2-microglobulina. Cuando se sospecha que hay anemia hemolítica autoinmunitaria, quizás sean útiles las pruebas de recuento de reticulocitos, bilirrubina indirecta, haptoglobina sérica, antiglobulina (Coombs directo) y crioaglutinina.
Citometría de flujo (para inmunofenotipificación).
Hibridación fluorescente in situ (FISH), (para identificar del(11q), del(13q), del(17p), trisomía 12 y t(11;14)).
Análisis de variantes de TP53.
Análisis de variantes de IGH.
Concentraciones de inmunoglobulinas séricas.
Pruebas del virus de inmunodeficiencia humana (VIH) y de la hepatitis B y C.
Por lo general, no se necesita una tomografía computarizada (TC) si no hay adenopatías periféricas. Cuando se identifican adenopatías generalizadas en el examen físico se debe investigar la presencia de adenopatías retroperitoneales.
Lo habitual es que no se necesite aspiración ni biopsia de médula ósea.
En este trastorno, el recuento de linfocitos en sangre a menudo supera o iguala 5000/mm3 de células que exhiben un inmunofenotipo característico (células B positivas para CD5 y DC23 [CD5+ y CD23+]).[6,7] A medida que se dispone de pruebas más sensibles para la detección en la sangre periférica de células B monoclonales parecidas a las de la LLC, los investigadores han logrado detectar linfocitosis monoclonal de células B en el 3 % de los adultos mayores de 40 años y en el 6 % de los adultos mayores de 60 años.[8] Es posible que dicha detección y diagnóstico tempranos indiquen una falsa mejora de la supervivencia en este grupo de pacientes, y quizás generen preocupaciones innecesarias o conlleven a tratamiento de algunos pacientes que permanecerían sin diagnóstico durante toda su vida, esta situación se conoce como sobrediagnóstico o pseudoenfermedad.[9,10]
La confusión con otras enfermedades a veces se evita mediante la determinación de los marcadores de superficie celular. Los linfocitos de la LLC coexpresan los antígenos CD19 y CD20 de las células B, y el antígeno CD5, que es propio de las células T.[11] Esta coexpresión solo sucede en otra enfermedad: el linfoma de células de manto. Las células B de la LLC expresan concentraciones relativamente bajas de inmunoglobulina de membrana (en comparación con las células B en sangre periférica normal) y una cadena ligera única (κ o λ).[12] La LLC se diagnostica cuando hay un incremento absoluto en la linfocitosis o hay infiltrado en la médula ósea que se acompañan de características morfológicas e inmunofenotípicas propias confirmatorias de una población clonal específica. En un análisis de una base de datos, casi todos los pacientes con diagnóstico de LLC exhibían clones de células B prediagnósticos en sangre periférica (en las muestras disponibles) hasta 77 meses antes del diagnóstico.[7,13]
Cerca del 1 % de los casos de LLC morfológicos expresan marcadores de células T (CD4 y CD7) y reordenamientos clonales de los genes del receptor de células T. Estos pacientes tienen una frecuencia más alta de lesiones cutáneas, linfocitos con formas variables y una mediana de supervivencia más corta (13 meses) con respuestas mínimas a la quimioterapia y a los inhibidores de los receptores de células B.[14]
El diagnóstico diferencial debe excluir las siguientes entidades:
Linfocitosis monoclonal de células B (LMB). La LMB, precursora de la LLC, se define como una población clonal de células B circulantes en sangre periférica de menos de 5 × 109 /l de células B, sin indicios de linfadenopatía o esplenomegalia.[15] La mayoría de los casos exhiben el inmunofenotipo de LLC. La incidencia de la LMB en la población general es del 5 % al 12 % y aumenta con la edad.[16] En las familias con 2 o más casos de LLC, la LMB tiene una prevalencia del 13 % al 18 %. La LMB con recuentos bajos (≤0,5 × 109 /l de células B) pocas veces progresa a una LLC sintomática, pero cuando hay recuentos altos a veces progresa a LLC sintomática a una tasa inferior al 2 % por año, incluso para casos familiares.[15,17] En 2 series seleccionadas con más de 900 pacientes a quienes se los controló de manera prospectiva durante una mediana de 5 a 7 años, el 7 % de los pacientes presentó una LLC sintomática que exigió tratamiento con quimioterapia.[8,18] En un estudio en el que se usó el Mayo Clinic Biobank se identificaron 1712 pacientes con LMB a partir de 10 139 muestras seleccionadas.[19] En el 95 % de estos pacientes se encontró recuento bajo de LMB. Al cabo de una mediana de seguimiento de 10 años, solo en el 0,58 % de los pacientes la enfermedad progresó a una neoplasia maligna linfoide.[19]
Macroglobulinemia de Waldenström. Esta afección es similar a la LLC en cuanto a la evolución natural y las opciones terapéuticas, excepto por el síndrome de hiperviscosidad relacionado con la macroglobulinemia que se produce por concentraciones elevadas de inmunoglobulina M.
Leucemia de linfocitos granulares grandes (LGG). La LGG se caracteriza por linfocitosis con un inmunofenotipo de linfocitos citotóxicos naturales (NK) (CD2, CD16 y CD56) o un inmunofenotipo de células T (CD2, CD3 y CD8).[20,21,22] Estos pacientes a menudo presentan neutropenia y antecedentes de artritis reumatoide. La evolución natural es de baja malignidad, a menudo marcada por anemia y esplenomegalia. Esta afección encaja en el grupo clínico del síndrome de Felty.[23] Un hallazgo genético característico en el 50 % de los pacientes con LGG de células T abarca las variantes patogénicas del gen STAT3.[24] Los pacientes sintomáticos con citopenias por lo general presentan células T positivas para CD-8 con receptores de superficie α o β y variante de STAT3, células T positivas para CD-8 con receptores de superficie γ o δ, o un fenotipo de células T NK mutado.[25,26] Por el contrario, los pacientes asintomáticos presentan células T positivas para CD-8 con STAT3 de tipo natural, células T positivas para CD4, y células NK de tipo natural.[25] El tratamiento incluye la administración oral de dosis bajas de ciclofosfamida o metotrexato, ciclosporina, y tratamiento de las infecciones bacterianas adquiridas durante la neutropenia grave.[20,22,27,28]
Para obtener información sobre la leucemia prolinfocítica, que antes se describía en este resumen, consultar la sección Tratamiento de la leucemia prolinfocítica de células T en Tratamiento del linfoma no Hodgkin periférico de células T.
Factores pronósticos
Los marcadores pronósticos ayudan a clasificar a los pacientes en los ensayos clínicos, también sirven para evaluar la necesidad de tratamiento y seleccionar el tipo de tratamiento.[2,29,30] Los factores pronósticos que quizás ayuden a predecir el desenlace clínico son el subgrupo citogenético, el estado mutacional de la inmunoglobulina y el inmunofenotipo de CD38.[2,31,32,33,34,35,36,37,38,39]
Los factores pronósticos son los siguientes:
Variante patogénica de IGH.[32,33,34,39,40] El hallazgo de un número significativo de variantes en esta región se relaciona con una mediana de supervivencia de más de 20 a 25 años. La ausencia de variantes se relaciona con una mediana de supervivencia de 8 a 10 años.
Resultados de la prueba de FISH. Las anomalías cromosómicas identificadas mediante FISH se asociaron con el pronóstico en estudios retrospectivos y prospectivos, y con el tiempo se ha observado que estas anomalías presentan una evolución clonal.[31,41,42,43] Se notificaron las siguientes anomalías cromosómicas:
del(13q) como marcador de pronóstico favorable (mediana de supervivencia general [SG] de 17 años en un estudio prospectivo).[43]
Trisomía 12 y del(11), que acarrean un pronóstico menos favorable (mediana de SG de 9 a 11 años en un estudio prospectivo).[43]
del(17p), que se relaciona con variantes patogénicas de TP53, tasa de respuesta precaria y duración corta de la respuesta a las opciones terapéuticas estándar.[39] del(17p), que se relaciona con el pronóstico más desfavorable (mediana de SG de 7 años en un ensayo prospectivo).[43,44,45]
La combinación de características citogenéticas anormales como las deleciones del(11q) o del(17p) (indicadoras de pronóstico más precario), y la negatividad de la proteína de 70 kDa asociada a la cadena ζ (indicador de mejor pronóstico) en el mismo paciente se tradujo en un pronóstico precario.[38]
Estos hallazgos ponen énfasis en la necesidad de estudios prospectivos de las combinaciones de estos marcadores pronósticos.[46]
Otros factores pronósticos son los siguientes:
Anemia y trombocitopenia. Estas son variables de pronóstico adverso importantes, pero solo si la LLC produce gran compromiso de la médula ósea. La anemia hemolítica autoinmunitaria y la púrpura trombocitopénica inmunitaria no confieren un pronóstico más precario.
Edad. La LLC se presenta principalmente en adultos de edad mediana y avanzada, el pronóstico empeora con la edad.[42]
Resultados de la tomografía por emisión de positrones con tomografía computarizada. Esta prueba solo se debe usar cuando hay fiebre recurrente, sudores nocturnos abundantes, pérdida de peso (>10 % del peso inicial en 6 meses), o ganglios linfáticos de crecimiento rápido, debido a que estos hallazgos quizás anuncien una transformación histológica a un linfoma difuso de células B grandes (LDCBG) (transformación de Richter). En una revisión retrospectiva de 432 pacientes, 209 pacientes exhibieron un valor de captación estandarizado máximo (SUVmáx.) de 5 o superior.[49] Entre estos pacientes, el 80 % presentó una LLC con características histológicas de gran malignidad o un síndrome de Richter, ambas entidades con pronósticos igualmente precarios. Cuando el SUVmáx. fue de 10 o superior, la tasa de SG a 5 años fue de solo un 30 %.[49]
Tiempo de duplicación linfocitario. Un tiempo de duplicación del recuento de glóbulos blancos de menos de 1 año acarrea un pronóstico más precario.[50]
β2-microglobulina. Las concentraciones más altas indican un pronóstico más precario.[51]
Transformación de Richter. En el 2 % al 10 % de los pacientes, la LLC se transformará en un linfoma de crecimiento rápido o más maligno, lo que se denomina transformación de Richter.[52] Por lo general, es un LDCBG del subtipo de células B activadas más agresivo. El pronóstico es similar al del LDCBG de novo en los casos en los que no se ha necesitado tratamiento para la LLC o cuando no hay una relación clonal entre la LLC y el LDCBG (que se identifica cuando alberga diferentes cadenas ligeras clonales o si se puede lograr la secuenciación en los sitios de recombinación VDJ [variable, diversidad, unión] en la región variable de la cadena pesada de inmunoglobulina).[52] Sin embargo, el pronóstico para la mayoría de los pacientes con transformación de Richter a LDCBG es precario (mediana de supervivencia, 6–14 meses) si la LLC ya se trató antes con quimioinmunoterapia,[53], inhibidores de la tirosina–cinasa de Bruton (BTK) o venetoclax.[54] No hay un abordaje terapéutico estándar para los pacientes con pronóstico precario. Los tratamientos en evaluación clínica incluyen la terapia de células T con receptor quimérico para el antígeno (CAR), los anticuerpos acopladores biespecíficos de células T y los inhibidores de BTK no covalentes.[52,55] A menudo se recomienda la consolidación con trasplante de células madre alogénicas si se logra una respuesta con la terapia de inducción.[52] En casos poco frecuentes de transformación de Richter de una LLC a un Linfoma de Hodgkin, el pronóstico es el mismo que para los pacientes con enfermedad de novo cuando se ajusta por edad.[56,57]
Eliminación de la enfermedad residual medible (ERM). La mejora de las tasas de respuesta a regímenes más intensivos ha mejorado la ERM. En un ensayo prospectivo con 493 pacientes, la eliminación de la ERM fue un factor de predicción independiente de la SG en un análisis multivariante.[58] El criterio indirecto de valoración de la eliminación de la enfermedad residual, tiene importancia pronóstica,[58,59] pero en un ensayo prospectivo aleatorizado no mejoró la supervivencia. Se necesita un estudio que incluya aquellos pacientes que no logran limpiar por completo la médula con el tratamiento de inducción y, luego, durante la recidiva estos pacientes se asignarían al azar a recibir un tratamiento alternativo versus el mismo tratamiento; se usaría la SG como criterio principal de valoración.[29,60]
Inmunofenotipo de CD38.[33,61] La positividad para CD38 (>30 %) se correlaciona con un pronóstico más precario, pero cuando se utiliza el estado mutacional de IGH como método de referencia para el pronóstico se encuentra una tasa de positivos falsos del 30 % y una tasa de negativos falsos del 50 %.
Otras neoplasias malignas. Los pacientes con LLC también tienen un riesgo alto de presentar otras neoplasias malignas, incluso antes del tratamiento.[62] Se hizo un análisis poblacional de casi 2 millones de pacientes oncológicos de la base de datos National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) Program. Se encontró que en los pacientes con LLC preexistente que después presentaron cáncer colorrectal y cáncer de mama, la supervivencia específica por cáncer fue significativamente inferior (cociente de riesgos instantáneos [CRI], 1,46; P < 0,001 del cáncer colorrectal y CRI, 1,41; P = 0,005 del cáncer de mama) que la supervivencia específica por cáncer en los pacientes con cáncer colorrectal y cáncer de mama sin antecedentes de LLC, incluso después de ajustar por edad, sexo, raza y estadio de la enfermedad, y de excluir las muertes relacionadas con la LLC.[63]
Un índice pronóstico internacional (IPI) para la LLC (LLC-IPI) permitió identificar 4 subgrupos pronósticos a partir del estado mutacional de IGH, el estadio clínico, la edad (≤65 vs. >65 años), y el estado de TP53 (sin anomalías vs. del(17p), variante de TP53 o ambas).[64] Mediante un sistema de clasificación para predecir el tiempo hasta el primer tratamiento de la LLC en estadio temprano, se identificaron tres factores de riesgo adverso: IGH sin mutación, recuento absoluto de linfocitos superior a 15 × 10 9 / l y ganglios linfáticos palpables.[65] Todos los modelos pronósticos nuevos, incluso el LLC-IPI que se usa a menudo, es posible que estén desactualizados debido al uso de terapias de primera línea muy efectivas, como los inhibidores de BCL2 y los inhibidores de BTK.[66] Será necesario revalidar dichos modelos pronósticos.
Seguimiento después del tratamiento
Las TC tienen una utilidad muy limitada para el seguimiento de los pacientes luego de completar el tratamiento. En solo 2 de 176 pacientes en 3 ensayos prospectivos del German CLL Study Group se decidió tratar la recaída a partir de los resultados de la TC o la ecografía.[67]
Referencias:
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Burger JA: Treatment of Chronic Lymphocytic Leukemia. N Engl J Med 383 (5): 460-473, 2020.
Anaissie EJ, Kontoyiannis DP, O'Brien S, et al.: Infections in patients with chronic lymphocytic leukemia treated with fludarabine. Ann Intern Med 129 (7): 559-66, 1998.
Mauro FR, Foa R, Cerretti R, et al.: Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood 95 (9): 2786-92, 2000.
Hallek M, Cheson BD, Catovsky D, et al.: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 131 (25): 2745-2760, 2018.
Hallek M, Cheson BD, Catovsky D, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12): 5446-56, 2008.
Shanafelt TD, Kay NE, Jenkins G, et al.: B-cell count and survival: differentiating chronic lymphocytic leukemia from monoclonal B-cell lymphocytosis based on clinical outcome. Blood 113 (18): 4188-96, 2009.
Rawstron AC, Bennett FL, O'Connor SJ, et al.: Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med 359 (6): 575-83, 2008.
Dighiero G: Monoclonal B-cell lymphocytosis--a frequent premalignant condition. N Engl J Med 359 (6): 638-40, 2008.
Fazi C, Scarfò L, Pecciarini L, et al.: General population low-count CLL-like MBL persists over time without clinical progression, although carrying the same cytogenetic abnormalities of CLL. Blood 118 (25): 6618-25, 2011.
DiGiuseppe JA, Borowitz MJ: Clinical utility of flow cytometry in the chronic lymphoid leukemias. Semin Oncol 25 (1): 6-10, 1998.
Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995.
Landgren O, Albitar M, Ma W, et al.: B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med 360 (7): 659-67, 2009.
Hoyer JD, Ross CW, Li CY, et al.: True T-cell chronic lymphocytic leukemia: a morphologic and immunophenotypic study of 25 cases. Blood 86 (3): 1163-9, 1995.
Shim YK, Rachel JM, Ghia P, et al.: Monoclonal B-cell lymphocytosis in healthy blood donors: an unexpectedly common finding. Blood 123 (9): 1319-26, 2014.
Slager SL, Lanasa MC, Marti GE, et al.: Natural history of monoclonal B-cell lymphocytosis among relatives in CLL families. Blood 137 (15): 2046-2056, 2021.
Shanafelt TD, Kay NE, Rabe KG, et al.: Brief report: natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J Clin Oncol 27 (24): 3959-63, 2009.
Slager SL, Parikh SA, Achenbach SJ, et al.: Progression and survival of MBL: a screening study of 10 139 individuals. Blood 140 (15): 1702-1709, 2022.
Semenzato G, Zambello R, Starkebaum G, et al.: The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood 89 (1): 256-60, 1997.
Lamy T, Loughran TP: How I treat LGL leukemia. Blood 117 (10): 2764-74, 2011.
Bowman SJ, Sivakumaran M, Snowden N, et al.: The large granular lymphocyte syndrome with rheumatoid arthritis. Immunogenetic evidence for a broader definition of Felty's syndrome. Arthritis Rheum 37 (9): 1326-30, 1994.
Koskela HL, Eldfors S, Ellonen P, et al.: Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366 (20): 1905-13, 2012.
Cheon H, Xing JC, Moosic KB, et al.: Genomic landscape of TCRαβ and TCRγδ T-large granular lymphocyte leukemia. Blood 139 (20): 3058-3072, 2022.
Barilà G, Grassi A, Cheon H, et al.: Tγδ LGLL identifies a subset with more symptomatic disease: analysis of an international cohort of 137 patients. Blood 141 (9): 1036-1046, 2023.
Loughran TP, Kidd PG, Starkebaum G: Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 84 (7): 2164-70, 1994.
Dhodapkar MV, Li CY, Lust JA, et al.: Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood 84 (5): 1620-7, 1994.
Developments in the treatment of lymphoproliferative disorders: rising to the new challenges of CLL therapy. A report of a symposium presented during the 48th American Society of Hematology Annual Meeting and Exposition, December 8, 2006, Orlando, Florida. Clin Adv Hematol Oncol 5 (3 Suppl 5): 1-14; quiz 15-6, 2007.
Pflug N, Bahlo J, Shanafelt TD, et al.: Development of a comprehensive prognostic index for patients with chronic lymphocytic leukemia. Blood 124 (1): 49-62, 2014.
Döhner H, Stilgenbauer S, Benner A, et al.: Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 343 (26): 1910-6, 2000.
Hamblin TJ, Davis Z, Gardiner A, et al.: Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 94 (6): 1848-54, 1999.
Damle RN, Wasil T, Fais F, et al.: Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 94 (6): 1840-7, 1999.
Rosenwald A, Alizadeh AA, Widhopf G, et al.: Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med 194 (11): 1639-47, 2001.
Klein U, Tu Y, Stolovitzky GA, et al.: Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med 194 (11): 1625-38, 2001.
Orchard JA, Ibbotson RE, Davis Z, et al.: ZAP-70 expression and prognosis in chronic lymphocytic leukaemia. Lancet 363 (9403): 105-11, 2004.
Rassenti LZ, Huynh L, Toy TL, et al.: ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med 351 (9): 893-901, 2004.
Kröber A, Bloehdorn J, Hafner S, et al.: Additional genetic high-risk features such as 11q deletion, 17p deletion, and V3-21 usage characterize discordance of ZAP-70 and VH mutation status in chronic lymphocytic leukemia. J Clin Oncol 24 (6): 969-75, 2006.
Byrd JC, Gribben JG, Peterson BL, et al.: Select high-risk genetic features predict earlier progression following chemoimmunotherapy with fludarabine and rituximab in chronic lymphocytic leukemia: justification for risk-adapted therapy. J Clin Oncol 24 (3): 437-43, 2006.
Kharfan-Dabaja MA, Chavez JC, Khorfan KA, et al.: Clinical and therapeutic implications of the mutational status of IgVH in patients with chronic lymphocytic leukemia. Cancer 113 (5): 897-906, 2008.
Kröber A, Seiler T, Benner A, et al.: V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood 100 (4): 1410-6, 2002.
Catovsky D, Fooks J, Richards S: Prognostic factors in chronic lymphocytic leukaemia: the importance of age, sex and response to treatment in survival. A report from the MRC CLL 1 trial. MRC Working Party on Leukaemia in Adults. Br J Haematol 72 (2): 141-9, 1989.
Shanafelt TD, Witzig TE, Fink SR, et al.: Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol 24 (28): 4634-41, 2006.
Grever MR, Lucas DM, Dewald GW, et al.: Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol 25 (7): 799-804, 2007.
Catovsky D, Richards S, Matutes E, et al.: Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 370 (9583): 230-9, 2007.
Binet JL, Caligaris-Cappio F, Catovsky D, et al.: Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 107 (3): 859-61, 2006.
Rai KR, Sawitsky A, Cronkite EP, et al.: Clinical staging of chronic lymphocytic leukemia. Blood 46 (2): 219-34, 1975.
Binet JL, Auquier A, Dighiero G, et al.: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48 (1): 198-206, 1981.
Falchi L, Keating MJ, Marom EM, et al.: Correlation between FDG/PET, histology, characteristics, and survival in 332 patients with chronic lymphoid leukemia. Blood 123 (18): 2783-90, 2014.
Montserrat E, Sanchez-Bisono J, Viñolas N, et al.: Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol 62 (3): 567-75, 1986.
Di Giovanni S, Valentini G, Carducci P, et al.: Beta-2-microglobulin is a reliable tumor marker in chronic lymphocytic leukemia. Acta Haematol 81 (4): 181-5, 1989.
Parikh SA, Kay NE, Shanafelt TD: How we treat Richter syndrome. Blood 123 (11): 1647-57, 2014.
Rossi D, Spina V, Deambrogi C, et al.: The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117 (12): 3391-401, 2011.
Kittai AS, Huang Y, Miller S, et al.: Outcomes of patients with Richter transformation without prior chemoimmunotherapy for CLL/SLL: an international multicenter retrospective study. [Abstract] Blood 142 (Suppl 1): A-497, 2023.
Kittai AS, Bond D, Huang Y, et al.: Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Richter Transformation: An International, Multicenter, Retrospective Study. J Clin Oncol 42 (17): 2071-2079, 2024.
Al-Sawaf O, Robrecht S, Bahlo J, et al.: Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35 (1): 169-176, 2021.
Stephens DM, Boucher K, Kander E, et al.: Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica 106 (11): 2845-2852, 2021.
Böttcher S, Ritgen M, Fischer K, et al.: Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol 30 (9): 980-8, 2012.
Strati P, Keating MJ, O'Brien SM, et al.: Eradication of bone marrow minimal residual disease may prompt early treatment discontinuation in CLL. Blood 123 (24): 3727-32, 2014.
Montserrat E, Moreno C, Esteve J, et al.: How I treat refractory CLL. Blood 107 (4): 1276-83, 2006.
Ghia P, Guida G, Stella S, et al.: The pattern of CD38 expression defines a distinct subset of chronic lymphocytic leukemia (CLL) patients at risk of disease progression. Blood 101 (4): 1262-9, 2003.
Tsimberidou AM, Wen S, McLaughlin P, et al.: Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol 27 (6): 904-10, 2009.
Solomon BM, Rabe KG, Slager SL, et al.: Overall and cancer-specific survival of patients with breast, colon, kidney, and lung cancers with and without chronic lymphocytic leukemia: a SEER population-based study. J Clin Oncol 31 (7): 930-7, 2013.
International CLL-IPI working group: An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol 17 (6): 779-90, 2016.
Condoluci A, Terzi di Bergamo L, Langerbeins P, et al.: International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood 135 (21): 1859-1869, 2020.
Kreuzberger N, Damen JA, Trivella M, et al.: Prognostic models for newly-diagnosed chronic lymphocytic leukaemia in adults: a systematic review and meta-analysis. Cochrane Database Syst Rev 7: CD012022, 2020.
Eichhorst BF, Fischer K, Fink AM, et al.: Limited clinical relevance of imaging techniques in the follow-up of patients with advanced chronic lymphocytic leukemia: results of a meta-analysis. Blood 117 (6): 1817-21, 2011.
Información sobre los estadios de la leucemia linfocítica crónica
No hay un sistema de estadificación estándar para la leucemia linfocítica crónica (LLC). El sistema de estadificación Rai (Cuadro 1) y la clasificación de Binet (Cuadro 2) se describen a continuación.[1,2] Un grupo de trabajo auspiciado por el Instituto Nacional del Cáncer (NCI) formuló directrices estandarizadas para determinar los criterios a partir de la aplicabilidad, la respuesta y los efectos tóxicos, con el fin de usarlos en los ensayos clínicos de LLC en el futuro.[3]
Sistema de estadificación Rai
Cuadro 1. Sistema de estadificación Rai
Estadio
Criterios de estadificación
Estadio 0
Linfocitosis absoluta (>15 000/mm3) sin adenopatías, hepatoesplenomegalia, anemia ni trombocitopenia.
Estadio I
Linfocitosis absoluta con linfadenopatías, sin hepatoesplenomegalia, anemia ni trombocitopenia.
Estadio II
Linfocitosis absoluta, con hepatomegalia o esplenomegalia, y linfadenopatías o sin estas.
Estadio III
Linfocitosis absoluta y anemia (hemoglobina <11 g/dl) con linfadenopatías, hepatomegalia, esplenomegalia o sin estas.
Estadio IV
Linfocitosis absoluta y trombocitopenia (<100 000/mm3) con linfadenopatías, hepatomegalia, esplenomegalia o anemia, o sin estas.
Clasificación de Binet
Cuadro 2. Sistema de clasificación de Binet
Estadio
Criterios de estadificación
a Las áreas linfoides son cervical, axilar, inguinal y esplénica.
Estadio clínico Aa
Ausencia de anemia o trombocitopenia y menos de 3 áreas de compromiso linfoide (estadios Rai 0, I, y II).
Estadio clínico Ba
Ausencia de anemia o trombocitopenia, y 3 o más áreas de compromiso linfoide (estadios Rai I y II).
Estadio clínico C
Anemia o trombocitopenia independientemente del número de áreas con hiperplasia linfoide (estadios Rai III y IV).
La estadificación de Binet integra el número de grupos ganglionares comprometidos por la enfermedad con la insuficiencia medular. El principal beneficio de esta clasificación se deriva del reconocimiento de una forma de la enfermedad con predominio esplénico que quizás acarrea un mejor pronóstico que lo considerado en la estadificación Rai, y el reconocimiento de que la presencia de anemia o trombocitopenia tiene un pronóstico similar y no amerita estadios separados. Ninguno de los sistemas divide las causas de citopenia en inmunitarias y no inmunitarias. Los pacientes con trombocitopenia o anemia, o ambos, como resultado de una infiltración extensa de la médula ósea con afectación de la producción celular (Rai III/IV, Binet C) tienen un pronóstico más precario que los pacientes con citopenias inmunitarias.[4]
Durante el International Workshop de LLC se recomendó la integración de los sistemas de clasificación Rai y Binet de la siguiente manera: A(0), A(I), A(II); B(I), B(II); y C(III), C(IV).[5] El grupo de trabajo auspiciado por el NCI publicó directrices para el diagnóstico y tratamiento de la LLC en el entorno de ensayos clínicos y de la práctica general.[3] El uso de estos sistemas permite la comparación de resultados clínicos y la formulación de directrices terapéuticas.
Referencias:
Rai KR, Sawitsky A, Cronkite EP, et al.: Clinical staging of chronic lymphocytic leukemia. Blood 46 (2): 219-34, 1975.
Binet JL, Auquier A, Dighiero G, et al.: A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 48 (1): 198-206, 1981.
Hallek M, Cheson BD, Catovsky D, et al.: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood 111 (12): 5446-56, 2008.
Moreno C, Hodgson K, Ferrer G, et al.: Autoimmune cytopenia in chronic lymphocytic leukemia: prevalence, clinical associations, and prognostic significance. Blood 116 (23): 4771-6, 2010.
Chronic lymphocytic leukemia: recommendations for diagnosis, staging, and response criteria. International Workshop on Chronic Lymphocytic Leukemia. Ann Intern Med 110 (3): 236-8, 1989.
Elección del tratamiento de la leucemia linfocítica crónica
El tratamiento de los pacientes con leucemia linfocítica crónica (LLC) se debe personalizar a partir del comportamiento clínico de la enfermedad.[1] A menudo se hace un tratamiento conservador porque esta enfermedad por lo general es incurable, se presenta en una población mayor y suele progresar muy lento.[2]
En los ensayos más antiguos de análisis de datos recabados desde los años 70 hasta los años 90, se observó que la mediana de supervivencia para todos los pacientes osciló de 8 a 12 años.[3,4] Sin embargo, con la adopción de los inhibidores del receptor de células B y la terapia dirigida a BCL2, la mediana de supervivencia de todos los pacientes no se ha alcanzado y el seguimiento supera los 10 años.
El tratamiento de los pacientes con LLC abarca observación, tratamiento de las complicaciones infecciosas, hemorrágicas o inmunológicas y varias opciones terapéuticas en monoterapia o combinación. En los pacientes asintomáticos, es posible diferir el tratamiento hasta que el paciente se torne sintomático conforme la enfermedad progresa.[3] Se requiere una observación rigurosa y citas frecuentes para controlar la evolución clínica porque la tasa de progresión varía entre pacientes, hay amplios períodos de estabilidad y algunas veces regresiones espontáneas.[5] Se necesita el seguimiento frecuente para evitar una progresión rápida, incluso en los pacientes asintomáticos que exhiben una del(17p) en el análisis de hibridación fluorescente in situ (FISH) (o aquellos con una variante patogénica de TP53). En un metanálisis de ensayos aleatorizados no se observó beneficio de supervivencia del uso de una terapia inmediata versus una terapia diferida en pacientes con enfermedad en estadio temprano.[6][Nivel de evidencia A1] En la mayoría de los pacientes con LLC progresiva el tratamiento no logra curar la enfermedad. Algunos pacientes que recibieron un trasplante alogénico de células madre lograron una supervivencia sin enfermedad (SSE) prolongada, en ocasiones de más de 20 años.[7,8,9,10,11] También se observó una SSE prolongada en pacientes más jóvenes (<60 años) que presentaban hipermutación en IGH y que recibieron el régimen de fludarabina, ciclofosfamida y rituximab (FCR).[12,13,14]
Los siguientes factores clínicos quizás sean útiles para predecir la progresión de la enfermedad:[2]
Variante patogénica de IGH.
Anomalías cromosómicas identificadas mediante análisis por FISH o citogenético.
β2-microglobulina.
Tiempo de duplicación linfocitario.
En el International Workshop on Chronic Lymphocytic Leukemia se definió la LLC sintomática o progresiva de la siguiente manera:[15]
Indicios de insuficiencia medular progresiva, es decir, inicio o empeoramiento de anemia o trombocitopenia (en algunos pacientes, recuentos de plaquetas <100 × 109 /l que permanecen estables durante un periodo prolongado, y no siempre exigen una intervención terapéutica inmediata). Un valor de hemoglobina inferior a 10 g/dl o recuentos de plaquetas inferiores a 50 × 109 /l por lo general indican la necesidad de tratamiento.
Esplenomegalia masiva (es decir, ≥6 cm debajo del margen costal izquierdo), progresiva o sintomática.
Linfadenopatías masivas (es decir, diámetro más largo ≥10 cm), progresivas o sintomáticas.
Linfocitosis progresiva con aumento del 50 % o superior durante un periodo de más de 2 meses, o tiempo de duplicación linfocitario (TDL) inferior a 6 meses. El TDL se calcula por regresión lineal mediante la extrapolación de los recuentos de linfocitos absolutos obtenidos cada 2 semanas durante un periodo de observación de 2 o 3 meses; los pacientes con recuentos linfocitarios sanguíneos iniciales inferiores a 30 × 109 /l quizás necesiten periodos de observación más largos para determinar el TDL. Se deben excluir factores que contribuyen a la linfocitosis diferentes a la LLC (por ejemplo, infecciones o administración de corticoesteroides).
Complicaciones autoinmunitarias, como anemia o trombocitopenia, con respuesta deficiente a los corticoesteroides.
Compromiso extraganglionar sintomático o funcional (por ejemplo, cutáneo, renal, pulmonar o raquídeo). Síntomas de la enfermedad definidos como cualquiera de las siguientes manifestaciones:
Pérdida de peso involuntaria de más del 10 % del peso en los 6 meses previos.
Cansancio intenso (es decir, puntaje de 2 o superior en la escala funcional del Eastern Cooperative Oncology Group, incapacidad laboral o incapacidad para desempeñar las actividades habituales).
Episodios de fiebre que superan 100,5°F o 38,0°C durante 2 o más semanas sin indicios de infección.
Sudores nocturnos por lo menos durante 1 mes sin indicios de infección.
Consideraciones para la elección del tratamiento
A continuación, se exponen los principios generales para establecer la secuencia de uso de las opciones terapéuticas disponibles:
Pese a que hay muchas opciones terapéuticas, los pacientes con LLC asintomáticos o con afectación mínima a menudo se observan fuera del ámbito de un ensayo clínico. El tratamiento suele comenzar cuando el paciente exhibe citopenias pronunciadas, o cuando la sintomatología afecta mucho la calidad de vida, por ejemplo, cuando hay linfadenopatías voluminosas en crecimiento o síntomas debilitantes.
Debido a que no se ha encontrado un tratamiento curativo diferente al trasplante, el objetivo inicial del tratamiento es potenciar la eficacia (mejora de la supervivencia general), con la menor toxicidad general posible, a corto y largo plazo.
La Administración de Alimentos y Medicamentos de los Estados Unidos aprobó el uso de los fármacos biológicos ibrutinib, acalabrutinib y venetoclax como primera línea para todos los pacientes con diagnóstico nuevo de LLC que requieran tratamiento.[16] En los pacientes con factores de pronóstico precario (en especial, aquellos con del(17p) o variantes patogénicas de TP53) se debe considerar el uso del ibrutinib, acalabrutinib o venetoclax.[17]
Los fármacos quimioterapéuticos estándar, como la fludarabina, la bendamustina, la ciclofosfamida y el clorambucilo producen daño en el DNA que se manifiestan como fenotipos más malignos y resistentes al tratamiento después de una recidiva, y es posible que induzcan neoplasias malignas secundarias. Sin embargo, se ha observado una SSE prolongada (superior a 10 años) con el uso del régimen FCR en pacientes más jóvenes (<60 años) con hipermutación en IGH.[12,13,14]
Evitar los alquilantes y los análogos de las purinas también previene las citopenias prolongadas y las infecciones recidivantes, persistentes y en ocasiones mortales que se observan después del tratamiento con estos fármacos.
El nuevo paradigma de terapia secuencial para la LLC incluye evitar al inicio el uso de fármacos quimioterapéuticos, siempre que sea posible.
Los pacientes de más edad con comorbilidades quizás toleren mejor el uso de los nuevos productos biológicos (como el ibrutinib o el venetoclax), una monoterapia de anticuerpos monoclonales (como dosis altas de rituximab) o una combinación de rituximab con fármacos quimioterapéuticos estándar en dosificaciones modificadas. En los pacientes de más edad (>65 años), la combinación de rituximab y bendamustina (régimen BR) produce menos efectos adversos y mejores desenlaces que el régimen FCR.[18]
Secuelas adversas de la enfermedad y el tratamiento
Las complicaciones infecciosas durante la enfermedad avanzada son, en parte, consecuencia de la hipogammaglobulinemia y de la incapacidad de lanzar la defensa humoral contra bacterias o virus. El herpes zóster causa una infección viral frecuente en estos pacientes, pero también hay infecciones por Pneumocystis carinii y Candida albicans. El reconocimiento temprano de las infecciones y el inicio de un tratamiento apropiado son elementos importantes de la supervivencia a largo plazo en estos pacientes. En un estudio aleatorizado sobre el uso de inmunoglobulina intravenosa (400 mg/kg cada 3 semanas por 1 año) en pacientes con LLC e hipogammaglobulinemia, se encontró un número significativamente inferior de infecciones bacterianas y se produjo un retraso significativo hasta el comienzo de la primera infección durante el período de estudio.[19] Sin embargo, no hubo efecto en la supervivencia. La administración intravenosa crónica de inmunoglobulina de manera rutinaria es costosa, y no se ha comprobado el beneficio a largo plazo (>1 año).[20,21]
En 2 informes retrospectivos, los pacientes con LLC que tuvieron que ser hospitalizados por COVID-19 antes de la inducción de las vacunas obtuvieron resultados precarios, con independencia del estadio.[22,23] En 1 de los estudios se observó un efecto protector de los inhibidores de la tirosina–cinasa de Bruton (BTK) (normalmente ibrutinib),[23] pero esto no se observó en el otro informe.[22] La tasa de letalidad de los pacientes con LLC se redujo del 35 %, a principios de 2020, al 11 %, a finales de 2020 y principios de 2021 (P < 0,001) debido a la mejora de las pruebas y la aparición de múltiples estrategias terapéuticas para la prevención y el tratamiento de la COVID-19.[22] En el caso de los pacientes que necesitan hospitalización, la tasa de letalidad se redujo del 40 % al 20 % (P = 0,003).
La anemia hemolítica autoinmunitaria o la trombocitopenia se presentan en pacientes con LLC en cualquier estadio.[24] El tratamiento inicial es corticoesteroides con alquilantes o sin estos (la fludarabina podría empeorar la anemia hemolítica). A menudo se necesita el uso de corticoesteroides para atenuar la destrucción autoinmunitaria, en lo posible antes de la quimioterapia mielosupresora, porque esto dificulta la administración exitosa de transfusiones de glóbulos rojos o plaquetas. Las alternativas de tratamiento son dosis altas de globulina, rituximab, ciclosporina, azatioprina, esplenectomía y dosis bajas de radioterapia dirigida al bazo.[3,25] El síndrome de lisis tumoral es una complicación infrecuente (1 de cada 300 pacientes) de la quimioterapia en pacientes que exhiben una enfermedad con gran masa tumoral.[26]
Un porcentaje pequeño de pacientes también presentan segundas neoplasias malignas y leucemias agudas a causa del tratamiento.[27] La transformación de la LLC en linfoma difuso de células B grandes (LDCBG) (conocido como síndrome de Richter) se produce en el 2 % al 10 % de los pacientes. Los factores de riesgo de transformación incluyen la ausencia de mutaciones en IGH, variantes patogénicas de TP53 o NOTCH1, pérdida de CDKN2A/B y cariotipo complejo.[28] Entre el 60 % y el 70 % de los pacientes presentan un LDCBG relacionado de forma clonal con el LLC y tienen un pronóstico significativamente más precario que los pacientes con LDCBG de novo.[29,30,31] Los pacientes con un LDCBG no relacionado de forma clonal con el LLC tienen un pronóstico mucho mejor, similar al del LDCBG de novo.[29] Sin embargo, la disponibilidad de secuenciación real de las cadenas pesadas de inmunoglobulina en la muestra original de LLC para compararla con la muestra transformada es limitada. Es posible que las firmas moleculares características sirvan como forma alternativa de evaluar el pronóstico.[32] Hasta el 20 % al 40 % de los pacientes con síndrome de Richter relacionado de forma clonal no presentan enfermedad más de 5 años después de recibir quimioterapia combinada intensiva, normalmente R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona) o Pola-R-CHP (polatuzumab, rituximab, ciclofosfamida, doxorrubicina y prednisona), a menudo seguida de un trasplante de células madre autógeno o alogénico.[33,34,35] Para obtener más información en inglés, consultar Aggressive B-Cell Non-Hodgkin Lymphoma Treatment. En dos informes retrospectivos, el LLC con transformación en linfoma de Hodgkin tuvo el mismo pronóstico que las presentaciones de novo de linfoma de Hodgkin a una edad equivalente.[36,37][Nivel de evidencia C3]
Los inhibidores de la BTK aumentaron el riesgo de hemorragia que requiriere hospitalización (riesgo a 3 años para los pacientes que recibieron ibrutinib, 8,8 % [intervalo de confianza (IC) 95 %, 6,5–11,7 %]) y de fibrilación auricular (incidencia a 3 años para los pacientes que recibieron ibrutinib, 22,7 % [IC 95 %, 19,0–26,6 %]).[38] En un ensayo aleatorizado con una mediana de seguimiento de 41 meses se observó menos fibrilación auricular en los pacientes con LLC que recibieron acalabrutinib en comparación con ibrutinib (9,4 vs. 16 %; P = 0,02).[39]
Gribben JG, O'Brien S: Update on therapy of chronic lymphocytic leukemia. J Clin Oncol 29 (5): 544-50, 2011.
Rozman C, Montserrat E: Chronic lymphocytic leukemia. N Engl J Med 333 (16): 1052-7, 1995.
Wierda WG, O'Brien S, Wang X, et al.: Prognostic nomogram and index for overall survival in previously untreated patients with chronic lymphocytic leukemia. Blood 109 (11): 4679-85, 2007.
Del Giudice I, Chiaretti S, Tavolaro S, et al.: Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood 114 (3): 638-46, 2009.
Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 91 (10): 861-8, 1999.
Ritgen M, Stilgenbauer S, von Neuhoff N, et al.: Graft-versus-leukemia activity may overcome therapeutic resistance of chronic lymphocytic leukemia with unmutated immunoglobulin variable heavy-chain gene status: implications of minimal residual disease measurement with quantitative PCR. Blood 104 (8): 2600-2, 2004.
Moreno C, Villamor N, Colomer D, et al.: Allogeneic stem-cell transplantation may overcome the adverse prognosis of unmutated VH gene in patients with chronic lymphocytic leukemia. J Clin Oncol 23 (15): 3433-8, 2005.
Khouri IF, Keating MJ, Saliba RM, et al.: Long-term follow-up of patients with CLL treated with allogeneic hematopoietic transplantation. Cytotherapy 4 (3): 217-21, 2002.
Doney KC, Chauncey T, Appelbaum FR, et al.: Allogeneic related donor hematopoietic stem cell transplantation for treatment of chronic lymphocytic leukemia. Bone Marrow Transplant 29 (10): 817-23, 2002.
Pavletic SZ, Khouri IF, Haagenson M, et al.: Unrelated donor marrow transplantation for B-cell chronic lymphocytic leukemia after using myeloablative conditioning: results from the Center for International Blood and Marrow Transplant research. J Clin Oncol 23 (24): 5788-94, 2005.
Thompson PA, Tam CS, O'Brien SM, et al.: Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 127 (3): 303-9, 2016.
Fischer K, Bahlo J, Fink AM, et al.: Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 127 (2): 208-15, 2016.
Rossi D, Terzi-di-Bergamo L, De Paoli L, et al.: Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 126 (16): 1921-4, 2015.
Hallek M, Cheson BD, Catovsky D, et al.: iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 131 (25): 2745-2760, 2018.
Burger JA, Tedeschi A, Barr PM, et al.: Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med 373 (25): 2425-37, 2015.
Cramer P, Tausch E, von Tresckow J, et al.: Durable remissions following combined targeted therapy in patients with CLL harboring TP53 deletions and/or mutations. Blood 138 (19): 1805-1816, 2021.
Eichhorst B, Fink AM, Bahlo J, et al.: First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 17 (7): 928-942, 2016.
Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. A randomized, controlled clinical trial. Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia. N Engl J Med 319 (14): 902-7, 1988.
Griffiths H, Brennan V, Lea J, et al.: Crossover study of immunoglobulin replacement therapy in patients with low-grade B-cell tumors. Blood 73 (2): 366-8, 1989.
Weeks JC, Tierney MR, Weinstein MC: Cost effectiveness of prophylactic intravenous immune globulin in chronic lymphocytic leukemia. N Engl J Med 325 (2): 81-6, 1991.
Mato AR, Roeker LE, Lamanna N, et al.: Outcomes of COVID-19 in patients with CLL: a multicenter international experience. Blood 136 (10): 1134-1143, 2020.
Scarfò L, Chatzikonstantinou T, Rigolin GM, et al.: COVID-19 severity and mortality in patients with chronic lymphocytic leukemia: a joint study by ERIC, the European Research Initiative on CLL, and CLL Campus. Leukemia 34 (9): 2354-2363, 2020.
Mauro FR, Foa R, Cerretti R, et al.: Autoimmune hemolytic anemia in chronic lymphocytic leukemia: clinical, therapeutic, and prognostic features. Blood 95 (9): 2786-92, 2000.
Kaufman M, Limaye SA, Driscoll N, et al.: A combination of rituximab, cyclophosphamide and dexamethasone effectively treats immune cytopenias of chronic lymphocytic leukemia. Leuk Lymphoma 50 (6): 892-9, 2009.
Cheson BD, Frame JN, Vena D, et al.: Tumor lysis syndrome: an uncommon complication of fludarabine therapy of chronic lymphocytic leukemia. J Clin Oncol 16 (7): 2313-20, 1998.
Maddocks-Christianson K, Slager SL, Zent CS, et al.: Risk factors for development of a second lymphoid malignancy in patients with chronic lymphocytic leukaemia. Br J Haematol 139 (3): 398-404, 2007.
Visentin A, Bonaldi L, Rigolin GM, et al.: The complex karyotype landscape in chronic lymphocytic leukemia allows the refinement of the risk of Richter syndrome transformation. Haematologica 107 (4): 868-876, 2022.
Rossi D, Spina V, Deambrogi C, et al.: The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood 117 (12): 3391-401, 2011.
Chigrinova E, Rinaldi A, Kwee I, et al.: Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood 122 (15): 2673-82, 2013.
Parry EM, Leshchiner I, Guièze R, et al.: Evolutionary history of transformation from chronic lymphocytic leukemia to Richter syndrome. Nat Med 29 (1): 158-169, 2023.
Parry EM, Ten Hacken E, Wu CJ: Richter syndrome: novel insights into the biology of transformation. Blood 142 (1): 11-22, 2023.
Robertson LE, Pugh W, O'Brien S, et al.: Richter's syndrome: a report on 39 patients. J Clin Oncol 11 (10): 1985-9, 1993.
Jain N, Keating M, Thompson P, et al.: Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med 380 (22): 2095-2103, 2019.
Ben-Dali Y, Hleuhel MH, da Cunha-Bang C, et al.: Richter's transformation in patients with chronic lymphocytic leukaemia: a Nationwide Epidemiological Study. Leuk Lymphoma 61 (6): 1435-1444, 2020.
Stephens DM, Boucher K, Kander E, et al.: Hodgkin lymphoma arising in patients with chronic lymphocytic leukemia: outcomes from a large multi-center collaboration. Haematologica 106 (11): 2845-2852, 2021.
Al-Sawaf O, Robrecht S, Bahlo J, et al.: Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia 35 (1): 169-176, 2021.
Abdel-Qadir H, Sabrie N, Leong D, et al.: Cardiovascular Risk Associated With Ibrutinib Use in Chronic Lymphocytic Leukemia: A Population-Based Cohort Study. J Clin Oncol 39 (31): 3453-3462, 2021.
Byrd JC, Hillmen P, Ghia P, et al.: Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J Clin Oncol 39 (31): 3441-3452, 2021.
Tratamiento de la leucemia linfocítica crónica asintomática
Opciones de tratamiento para la leucemia linfocítica crónica asintomática
Observación
Debido a la naturaleza poco activa de la leucemia linfocítica crónica (LLC) en pacientes asintomáticos o con afectación mínima, no se aconseja el uso de quimioterapia y el abordaje más aceptado es la observación.[1] Se requieren controles frecuentes con observación cuidadosa para vigilar la evolución clínica porque la tasa de progresión varía con largos períodos de estabilidad y, algunas veces, regresiones espontáneas. Un nomograma que predice el tiempo hasta el primer tratamiento se basa en el número de sitios de ganglios linfáticos, el tamaño de los ganglios linfáticos cervicales, la concentración de lactato-deshidrogenasa, el estado mutacional de IGH y la presencia de la del(11q) o la del(17p) determinadas mediante análisis de hibridación fluorescente in situ.[2] La regresión espontánea que se manifiesta por una reducción sostenida del clon maligno sin tratamiento, se observa en menos del 5 % de los pacientes. Estos pacientes tienen casi exclusivamente una hipermutación en IGH.[3]
Evidencia (observación):
El French Cooperative Group on Chronic Lymphocytic Leukemia asignó al azar a 1535 pacientes con enfermedad en estadio A que no se trató antes, a recibir clorambucilo o ningún tratamiento inmediato.[4]
Los resultados no indicaron beneficio para la supervivencia del tratamiento inmediato con clorambucilo.[4][Nivel de evidencia A1]
En un metanálisis se evaluaron 6 ensayos de pacientes con LLC en estadio temprano en los que se usó terapia inmediata versus terapia diferida con clorambucilo (entre estos ensayos, el del French Cooperative Group mencionado antes).[5]
Los resultados no indicaron diferencia en la supervivencia general a 10 años.[5][Nivel de evidencia A1]
Pese a la existencia de muchas opciones terapéuticas, se debe considerar la observación en los pacientes asintomáticos o con afectación mínima, incluso si tienen factores de pronóstico adverso. El tratamiento se inicia cuando los pacientes exhiben citopenias pronunciadas o los síntomas deterioran la calidad de vida.
No se han publicado resultados de ensayos clínicos en los que se corrobore la superioridad del tratamiento inmediato con inhibidores del receptor de células B o inhibidores BCL2 en comparación con la observación en los pacientes asintomáticos o con afectación mínima.
Antes de dejar de lado la observación o la conducta expectante, se necesitan ensayos clínicos para establecer si los desenlaces mejoran cuando se usan las nuevas terapias biológicas en pacientes asintomáticos.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Casper JT: Prognostic features of early chronic lymphocytic leukaemia. International Workshop on CLL. Lancet 2 (8669): 968-9, 1989.
Molica S, Giannarelli D, Gentile M, et al.: External validation on a prospective basis of a nomogram for predicting the time to first treatment in patients with chronic lymphocytic leukemia. Cancer 119 (6): 1177-85, 2013.
Kwok M, Oldreive C, Rawstron AC, et al.: Integrative analysis of spontaneous CLL regression highlights genetic and microenvironmental interdependency in CLL. Blood 135 (6): 411-428, 2020.
Dighiero G, Maloum K, Desablens B, et al.: Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med 338 (21): 1506-14, 1998.
Chemotherapeutic options in chronic lymphocytic leukemia: a meta-analysis of the randomized trials. CLL Trialists' Collaborative Group. J Natl Cancer Inst 91 (10): 861-8, 1999.
Tratamiento de la leucemia linfocítica crónica sintomática o progresiva
Opciones de tratamiento para la leucemia linfocítica crónica sintomática o progresiva
A continuación, se indican los regímenes que se consideran abordajes de tratamiento de primera línea en pacientes con leucemia linfocítica crónica (LLC) que presentan progresión sintomática:
R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona) (solo si hay sospecha clínica de síndrome de Richter con progresión histológica).
Estos abordajes se compararon en varios ensayos clínicos prospectivos. En la mayoría de los pacientes se prefiere un abordaje de tratamiento de primera línea sin quimioterapia, pero esta es preceptiva en los pacientes con enfermedad positiva para del(17p) o una mutación en TP53.[1,2,3,4,5]
Inhibidores de la BTK
Ibrutinib versus zanubrutinib
Evidencia (ibrutinib vs. zanubrutinib):
En un ensayo prospectivo aleatorizado de 652 pacientes con LLC en recaída o resistente al tratamiento se comparó el ibrutinib y el zanubrutinib.[6]
Al cabo de una mediana de seguimiento de 29,6 meses, la tasa de supervivencia sin progresión (SSP) a 2 años fue del 78,4 % en el grupo del zanubrutinib y del 65,9 % en el grupo del ibrutinib (cociente de riesgos instantáneos [CRI], 0,65; intervalo de confianza [IC] 95 %, 0,49–0,86; P = 0,002).[6][Nivel de evidencia B1]
Se produjeron trastornos cardíacos que llevaron a la interrupción del tratamiento en 1 paciente (0,3 %) del grupo del zanubrutinib y en 14 pacientes (4,3 %) del grupo del ibrutinib. Las 6 muertes producidas como consecuencia de episodios cardiacos se produjeron en pacientes que recibieron ibrutinib.
Ibrutinib versus acalabrutinib
Evidencia (ibrutinib vs. acalabrutinib):
En un ensayo prospectivo aleatorizado de 533 pacientes sin tratamiento previo se comparó los inhibidores de la BTK acalabrutinib e ibrutinib.[7]
Al cabo de una mediana de seguimiento de 41 meses, el acalabrutinib no fue inferior al ibrutinib, la mediana de SSP fue de 38,4 meses para los pacientes de los dos grupos del ensayo (CRI, 1,00; IC 95 %, 0,79–1,27).[7][Nivel de evidencia B1]
La incidencia de fibrilación auricular de cualquier grado fue menor en los pacientes que recibieron el acalabrutinib que en los que recibieron el ibrutinib (9,4 vs. 16,0 %, P = 0,02). En este ensayo, la incidencia de diarrea y cefaleas fue significativamente mayor en los pacientes que recibieron acalabrutinib (P < 0,05), mientras que la incidencia del dolor osteomuscular fue mayor en los pacientes que recibieron ibrutinib (P = 0,0229).[8] Las arritmias ventriculares y las muertes súbitas parecen ser un efecto secundario de todos los inhibidores de la BTK; en el caso del acalabrutinib, estos eventos se producen con un riesgo relativo de 8,2 con respecto a los controles emparejados por edad.[9]
Ibrutinib versus ibrutinib y rituximab versus bendamustina y rituximab
Evidencia (ibrutinib vs. ibrutinib y rituximab vs. bendamustina y rituximab):
En un ensayo prospectivo, se asignaron al azar 547 pacientes de 65 años o más sin tratamiento previo a un grupo de bendamustina y rituximab (BR), un grupo de ibrutinib solo o un grupo de ibrutinib y rituximab.[10]
Al cabo de una mediana de seguimiento de 38 meses, la tasa de SSP a 2 años fue del 74 % para los pacientes que recibieron el régimen BR. Las tasas fueron significativamente más altas para los pacientes que recibieron ibrutinib solo (87 %; CRI, 0,39; IC 95 %, 0,25–0,58) o ibrutinib y rituximab (88 %; CRI, 0,38; IC 95 %, 0,25–0,59; P < 0,001).[10][Nivel de evidencia B1]
No hubo ninguna diferencia en la SSP de los 2 grupos que recibieron el ibrutinib (CRI, 1,00; IC 95 %, 0,62–1,62; P = 0,49) ni en la supervivencia general (SG) de todos los grupos.
Ibrutinib versus rituximab e ibrutinib
Evidencia (ibrutinib vs. rituximab e ibrutinib):
En un ensayo aleatorizado prospectivo participaron 208 pacientes sin tratamiento previo o con enfermedad en recaída. Los pacientes recibieron rituximab e ibrutinib o ibrutinib solo.[11]
Al cabo de una mediana de seguimiento de 36 meses, no hubo diferencia en la SSP (86 %; CRI, 1,04; IC 95 %, 0,49−2,20; P = 0,91).[11][Nivel de evidencia B1]
Ibrutinib versus fludarabina, ciclofosfamida y rituximab
El ibrutinib es un inhibidor selectivo irreversible de la BTK, una molécula de señalización que participa en la primera parte de la cadena de señalización del receptor de células B.
Evidencia (ibrutinib vs. fludarabina, ciclofosfamida y rituximab):
En el ensayo prospectivo y aleatorizado ECOG-E1912 (NCT02048813) se comparó ibrutinib y rituximab con el régimen fludarabina, ciclofosfamida y rituximab (FCR). Se asignó al azar, en una proporción 2:1, a 529 pacientes con LLC que no recibieron tratamiento previo para recibir ibrutinib y rituximab, seguidos de mantenimiento con ibrutinib (354 pacientes) o 6 ciclos de FCR (175 pacientes).[12,13]
Al cabo de una mediana de seguimiento de 5,8 años, la tasa de SG a 5 años favoreció al grupo de ibrutinib (95 vs. 89 %) (CRI, 0,47; IC 95 %, 0,25–0,89; P = 0,018).[13][Nivel de evidencia A1]
En los 281 pacientes sin variantes patogénicas de IGH, la tasa de SSP a 5 años favoreció el uso de ibrutinib y rituximab, versus el régimen FCR (75 vs. 33 %) (CRI, 0,27; IC 95 %, 0,18−0,41; P < 0,0001). En los 114 pacientes sin variantes patogénicas de IGH, la tasa de SSP a 5 años también fue significativamente diferente entre los 2 grupos, siendo del 83 % en el grupo de ibrutinib y del 68 % en el grupo de FCR (CRI, 0,27; IC 95 %, 0,11−0,62; P = 0,001).[12,13,14]
Aunque la enfermedad residual medible (ERM) indetectable fue inferior al 10 % entre los 12 y 36 meses de seguimiento, los pacientes con ERM detectable no tuvieron una SSP significativamente más precaria (P = 0,14 a los 12 meses, P = 0,90 a los 24 meses y P = 0,53 a los 36 meses).[15]
Zanubrutinib versus bendamustina y rituximab
Evidencia (zanubrutinib vs. bendamustina y rituximab):
En un ensayo prospectivo aleatorizado participaron 590 pacientes de 65 años o más no tratados previamente. Los pacientes se asignaron al azar a recibir zanubrutinib o bendamustina y rituximab.[16]
Al cabo de una mediana de seguimiento de 26,2 meses, la tasa de SSP a 2 años fue del 85,5 % (IC 95 %, 80,1–89,6 %) para los pacientes que recibieron zanubrutinib y del 69,5 % (IC 95 %, 62,4–75,5 %) para los pacientes que recibieron BR (CRI, 0,42; IC 95 %, 0,28–0,63; P < 0,0001).[16][Nivel de evidencia B1]
Acalabrutinib y obinutuzumab versus acalabrutinib versus clorambucil y obinutuzumab
El acalabrutinib es un inhibidor covalente irreversible y muy selectivo de la BTK que se diseñó para reducir la toxicidad gastrointestinal y el riesgo de fibrilación atrial del ibrutinib.
Evidencia (acalabrutinib y obinutuzumab vs. acalabrutinib vs. clorambucil y obinutuzumab):
En el ensayo prospectivo ELEVATE TN (NCT02475681) se incluyeron 535 pacientes de 65 años o más sin tratamiento previo y con comorbilidades (por ejemplo, depuración de creatinina <70 ml/min). Los pacientes se asignaron al azar a 3 grupos: acalabrutinib y obinutuzumab, acalabrutinib solo, o clorambucilo y obinutuzumab.[17]
Al cabo de una mediana de seguimiento de 28 meses, las tasas de SSP a 24 meses fueron del 93 % para el acalabrutinib y el obinutuzumab (CRI, 0,10; IC 95 %, 0,06−0,17; P < 0,0001), del 87 % para el acalabrutinib solo (CRI, 0,20; IC 95 %, 0,13−0,30; P < 0,0001) y del 47 % para el clorambucilo y el obinutuzumab.[17][Nivel de evidencia B1]
Hubo una diferencia pequeña pero significativa en las tasas de SSP de los 2 grupos de acalabrutinib (93 vs. 87 % a los 24 meses) a favor de la combinación (CRI, 0,49; IC 95 %, 0,26−0,95).[17]
Venetoclax con uso inicial de obinutuzumab o rituximab
Venetoclax y obinutuzumab versus clorambucilo y obinutuzumab
Evidencia (venetoclax y obinutuzumab vs. clorambucilo y obinutuzumab):
En el ensayo prospectivo CLL14 (NCT02242942) se asignaron al azar a 432 pacientes sin tratamiento previo que tenían comorbilidad importante (puntaje de 6 o superior en la Cumulative Illness Rating Scale; mediana de edad, 72 años) para recibir venetoclax (inhibidor muy selectivo de BCL2) y obinutuzumab (anticuerpo monoclonal humano anti-CD20) versus clorambucilo y obinutuzumab.[18]
Al cabo de una mediana de seguimiento de 28,1 meses, la tasa de SSP a 2 años fue significativamente más alta para los pacientes que recibieron venetoclax con obinutuzumab, del 88,2 % (IC 95 %, 83,7‒92,6 %), en comparación con el clorambucilo y obinutuzumab, del 64,1 % (IC 95 %, 57,4‒70,8 %) (CRI, 0,35; IC 95 %, 0,23‒0,53; P < 0,001).[18][Nivel de evidencia B1]
Después de una mediana de seguimiento de 52,4 meses, la tasa de SSP a 4 años para el grupo de venetoclax fue del 74 % y la ERM indetectable en la médula ósea a 3 años fue del 57 % (<10-4) versus una tasa de SSP a 4 años para el grupo de clorambucilo del 35,4 % y una ausencia de ERM en la médula ósea a 3 años del 17 % (CRI, 0,33; IC 95 %, 0,25−0,45; P < 0,0001).[19,20][Nivel de evidencia B1]
Venetoclax y rituximab versus bendamustina y rituximab
Evidencia (venetoclax y rituximab versus bendamustina y rituximab):
En el ensayo prospectivo MURANO (NCT02005471) se incluyeron 389 pacientes con LLC en recaída o resistente al tratamiento. Los pacientes se asignaron al azar para recibir venetoclax y rituximab (VenR) (venetoclax durante 2 años con rituximab durante los primeros 6 meses) versus bendamustina y rituximab (BR) durante 6 meses.
Al cabo de una mediana de seguimiento de 24 meses, la tasa de SSP a 2 años fue del 84,9 % para VenR y del 36,3 % para BR (CRI, 0,17; IC 95 %, 0,11–0,25; P < 0,001).[21,22] En una actualización publicada como resumen, al cabo de una mediana de seguimiento de 48 meses, se observó una tasa de SG a 4 años del 85,3 % para VenR y del 66,8 % para BR (CRI, 0,41; IC 95 %, 0,26−0,65; P < 0,0001).[23][Nivel de evidencia A1]
Al cabo de una mediana de seguimiento de 59,2 meses, la SG a 5 años fue del 82,1 % (IC 95 %, 76,4–87,8 %) en los pacientes que recibieron VenR y del 62,2 % (IC 95 %, 54,8–69,6 %) en los pacientes que recibieron BR (P < 0,0001).[24][Nivel de evidencia A1] Para VenR, la mediana de tiempo hasta el siguiente tratamiento fue de 57,8 meses (IC 95 %, 55,1–no estimable).
En el 43 % de los pacientes del grupo VenR que alcanzaron una ERM indetectable al final del tratamiento, la mediana de tiempo hasta la conversión a ERM fue de 19,4 meses. La mediana de tiempo desde la conversión a ERM hasta la enfermedad clínica progresiva manifiesta fueron otros 25,2 meses.[24]
Bendamustina y rituximab
Bendamustina y rituximab versus fludarabina, ciclofosfamida y rituximab
Evidencia (bendamustina y rituximab versus fludarabina, ciclofosfamida y rituximab):
El German CLL Study Group comparó el uso de bendamustina y rituximab (BR) versus fludarabina, ciclofosfamida y rituximab (FCR) como terapia de primera línea en pacientes con LLC que necesitaban tratamiento.[25]
Después de una mediana de seguimiento de 37,1 meses, la mediana de SSP fue mejor para los pacientes que recibieron FCR (55,2 vs. 41,7 meses; CRI, 1,64; IC 90 %, 1,31–2,06, P = 0,001), pero no hubo diferencia en la tasa de SG a 3 años (91 vs. 92 %, no significativa).[25][Nivel de evidencia B1]
En los pacientes mayores de 65 años, no hubo ninguna diferencia en la SSP entre los 2 grupos, pero se produjeron más infecciones en el grupo de FCR que en el de BR (infección de grado 3 a 5, 47 vs. 27 %).
Fludarabina, ciclofosfamida y rituximab
Fludarabina, ciclofosfamida y rituximab
El régimen de fludarabina, ciclofosfamida y rituximab (FCR) se usa en pacientes con hipermutación en IGH.
Evidencia (FCR):
En varios ensayos se usó FCR para los pacientes adecuados con variantes patogénicas de IGH que necesitaban tratamiento.
La tasa de SSP superó el 60 % al cabo de más de 10 años.[26,27,28][Nivel de evidencia C2] No obstante, se observaron recaídas tardías después de 10 años.
Inhibidor de la tirosina–cinasa de Bruton y venetoclax
Inhibidor de la tirosina–cinasa de Bruton (ibrutinib o acalabrutinib) y venetoclax
Evidencia (inhibidor de la tirosina–cinasa de Bruton [ibrutinib o acalabrutinib] y venetoclax):
En un ensayo prospectivo aleatorizado se incluyó a 523 pacientes con LLC que no habían recibido tratamiento previo. Los pacientes recibieron ibrutinib y venetoclax durante un máximo de 6 años, o FCR durante 6 ciclos.[29] Esta comparación se incluyó en una aleatorización más amplia de 3 grupos, que también incluía ibrutinib solo durante un máximo de 6 años; se consideró que las estadísticas eran demasiado preliminares para analizar el grupo de ibrutinib solo versus los otros grupos.
Al cabo de una mediana de seguimiento de 43,7 meses, la tasa de SG a 3 años fue del 98,0 % (IC 95 %, 95,2–99,2 %) en el grupo de ibrutinib y venetoclax y del 93,0 % (IC 95 %, 88,9–95,6 %) en el grupo de FCR (CRI, 0,31; IC 95 %, 0,15–0,67).[29][Nivel de evidencia A1]
El beneficio en SG a 3 años favoreció al grupo de ibrutinib y venetoclax (CRI, 0,23; IC 95 %, 0,06–0,81) en los pacientes sin variantes patogénicas de IGH, pero no en aquellos con variantes patogénicas de IGH (CRI, 0,61; IC 95 %, 0,20–1,82).
La tasa de SSP a 3 años fue del 97,2 % (IC 95 %, 94,1–98,6 %) en el grupo de ibrutinib y venetoclax y del 76,8 % (IC 95 %, 70,8–81,7 %) en el grupo de FCR (CRI, 0,13; IC 95 %, 0,07–0,24; P < 0,001).
La negatividad de la ERM fue un requisito para interrumpir el tratamiento con venetoclax e ibrutinib después de 2 años. En este ensayo no se asignó al azar a los pacientes a recibir tratamiento continuado o a la interrupción del tratamiento en función del estado de la ERM. La utilidad y la importancia de la ERM no pudieron determinarse a partir de este ensayo debido a su diseño.
En un ensayo prospectivo aleatorizado (GLOW [NCT03462719]) se incluyó a 211 pacientes con LLC que no recibieron tratamiento previo. Los pacientes tenían 65 años o más, o entre 18 y 64 años con comorbilidades y un estado funcional de 0 a 2 según el Eastern Cooperative Oncology Group. Los pacientes se asignaron al azar a recibir ibrutinib y venetoclax de duración fija (15 meses en total) o clorambucilo y obinutuzumab (6 meses en total).[30]
Al cabo de una mediana de seguimiento de 46 meses, la tasa de SSP a 42 meses fue del 74,6 % en el grupo de ibrutinib y venetoclax y del 24,8 % en el grupo de clorambucilo y obinutuzumab (CRI, 0,214; IC 95 %, 0,138–0,334; P < 0,0001).[30][Nivel de evidencia B1]
El ibrutinib se administró durante 3 meses antes de la combinación de venetoclax con ibrutinib para evitar el síndrome de lisis tumoral.
A los 3 meses de finalizar el tratamiento, el 40,6 % de los pacientes que recibieron ibrutinib y venetoclax lograron una ERM indetectable (<10-5 mediante secuenciación de última generación) en la médula ósea, y el 43,4 % de los pacientes lograron una ERM indetectable en la sangre periférica.[30] Sin embargo, la tasa de SSP superó el 90 % a los 12 meses de finalizar el tratamiento, con independencia de si la ERM era detectable o no.
En un ensayo de fase II, 80 pacientes sin tratamiento previo, de 65 años o más o que tenían una enfermedad de riesgo alto, recibieron tratamiento con ibrutinib durante 3 meses, seguido de ibrutinib y venetoclax durante 24 meses en total.[31]
Al cabo de una mediana de seguimiento de 38,5 meses, la tasa de respuesta a 2 años fue del 82 %, la tasa de SSP a 3 años fue del 93 % (IC 95 %, 88–99 %) y la tasa de SG a 3 años fue del 96 %.[31][Nivel de evidencia C1]
La toxicidad de la combinación es similar a la toxicidad de cada medicamento en monoterapia, además se evita el síndrome de lisis tumoral propio del inicio del tratamiento con venetoclax.
Las tasas altas de ERM indetectable (<10-4 en la prueba de citometría de flujo de 8 colores) en sangre periférica (75 %) y en la médula ósea (72 %) no tienen precedente. La importancia clínica de este hallazgo se debe analizar en ensayos prospectivos aleatorizados para establecer los desenlaces clínicos, en comparación con el uso de cada medicamento por separado. Esta combinación también se ha probado en el entorno de recaída o resistencia al tratamiento.[32]
En un ensayo de fase II participaron 159 pacientes de 70 años o más que no recibieron tratamiento previo. Los pacientes recibieron 3 ciclos de ibrutinib solo, seguidos de 12 ciclos de ibrutinib y venetoclax.[33]
Al cabo de una mediana de seguimiento de 27,9 meses, la tasa de respuesta completa fue del 55 % (IC 95 %, 48–63 %), la tasa de ERM indetectable fue del 77 % (IC 95 %, 70–83 %) en sangre y del 60 % (IC 95 %, 52–67 %) en médula ósea, la tasa de SSP a 2 años fue del 95 % (IC 95 %, 90–97 %) y la tasa de SG a 2 años fue del 98 % (IC 95 %, 94–99 %).[33][Nivel de evidencia C1]
La toxicidad de la combinación ibrutinib-venetoclax fue similar a la de cualquiera de los dos fármacos por separado y el ibrutinib inicial en monoterapia evitó el síndrome de lisis tumoral.
En un ensayo de fase II participaron 41 pacientes sin tratamiento previo que recibieron ibrutinib con venetoclax y obinutuzumab. Los pacientes con ERM indetectable en el ciclo 16 tenían la opción de interrumpir el tratamiento si presentaban una respuesta completa.[34]
Al cabo de una mediana de seguimiento de 38,4 meses, la tasa de SSP a 3 años fue del 79,9 % y la tasa de SG a 3 años fue del 92,6 %.[34][Nivel de evidencia C1]
En resumen, en estos estudios se estableció el uso del venetoclax con obinutuzumab o rituximab, o el uso del ibrutinib, el acalabrutinib o el zanubrutinib, como terapia de primera línea en pacientes de LLC que no recibieron tratamiento previo. Una menor tasa de fibrilación auricular tal vez favorece el uso del acalabrutinib o el zanubrutinib frente al ibrutinib.[6,7,35,36] A diferencia del ibrutinib o del acalabrutinib, que se administran de manera continua hasta la recaída, es posible suspender el venetoclax al cabo de 12 meses, y mantener una remisión duradera. Cuando es necesario, el venetoclax, el ibrutinib, el acalabrutinib o el zanubrutinib se administran de nuevo y de manera exitosa.[36,37] Estos fármacos dirigidos también son eficaces en pacientes con variantes patogénicas de TP53.[38] Los diferentes regímenes de estos medicamentos, como en monoterapia o en combinación, con obinutuzumab o sin este, se deben evaluar en ensayos prospectivos aleatorizados. Varios ensayos provocativos de fase II y fase III con estas combinaciones han dado lugar a tasas sin precedentes de enfermedad sin ERM que parecen más duraderas;[31,39,40,41,42] para saber si esto se traduce en un beneficio clínico respecto a un abordaje más secuencial son necesarios ensayos prospectivos aleatorizados. Estas combinaciones acarrean bastante toxicidad financiera, lo que exige verificación de la superioridad en la eficacia. Estos ensayos justifican aún más el uso de un abordaje sin quimioterapia durante la terapia de primera línea de la LLC en lugar del estándar anterior de usar el régimen BR y el régimen FCR (que probaron ser más eficaces que los regímenes de clorambucilo). Los pacientes con mayor riesgo de recaída presentan múltiples factores de pronóstico precario, como variantes patogénicas de TP53, deshidrogenasa láctica (LDH) elevada, microglobulina beta-2 elevada y tratamiento previo.[43] Estos pacientes de mayor riesgo deben considerar la participación en ensayos clínicos.
Evaluación de la enfermedad residual medible fuera del entorno de un ensayo clínico
La ERM indetectable (células de LLC en muestras aspiradas de sangre periférica o médula ósea, ≤1 × 10-4) puede confirmarse mediante citometría de flujo o secuenciación de última generación. El logro de una ERM indetectable indica una remisión completa más rigurosa , la cual funcionó como factor pronóstico de la mejora de la SSP y la SG en muchos estudios.[20,24,44,45] El uso de una terapia de tiempo limitado no es tan valioso en la práctica clínica habitual; su uso con venetoclax, o la combinación en investigación de venetoclax y un inhibidor de la tirosina–cinasa de Bruton, han producido respuestas completas, e incluso ERM indetectable, en la mayoría de los pacientes.[24,44,46]
La evaluación de la ERM indetectable se ha convertido en un parámetro estándar para definir las respuestas en todos los ensayos clínicos modernos de LLC. La ERM indetectable tiene valor pronóstico, pero no está clara su condición de marcador predictivo. El valor potencial de las pruebas de ERM en la práctica clínica habitual depende de si se puede utilizar para la toma de decisiones clínicas, como suspender, cambiar o continuar el tratamiento. Para obtener evidencia contundente sobre esta intervención se necesitaría un ensayo clínico aleatorizado prospectivo en el que se use la ERM como biomarcador predictivo para un grupo que logre una ventaja en la SG, en comparación con un grupo de control que no tenga en cuenta el estado de la ERM. Dicha evidencia aún no se ha obtenido. La SSP mejorada en pacientes con LLC no predice de manera directa la SG, de manera similar a los resultados observados en el linfoma folicular y otras neoplasias linfoides de crecimiento lento.
En un ensayo de fase II se usó una combinación de venetoclax e ibrutinib en pacientes con ERM indetectable después de 1 año de tratamiento. Los pacientes se asignaron al azar a recibir terapia de mantenimiento con ibrutinib o interrupción del tratamiento.[46] Todos los pacientes positivos para ERM siguieron recibiendo ibrutinib. Al cabo de una mediana de seguimiento de 34,4 meses, las tasas de SSP a 1 año fueron del 98 % en los pacientes con ERM indetectable que interrumpieron el tratamiento, del 96 % en los pacientes con ERM indetectable que continuaron recibiendo ibrutinib y del 97 % en los pacientes con ERM positiva que siguieron recibiendo ibrutinib. Todos los pacientes evolucionaron bien a los 12 meses. Conocer el estado de la ERM para indicar la suspensión del tratamiento no influyó en el desenlace.[46]
En otro ensayo de fase II se incluyó a 70 pacientes no tratados previamente que recibieron 1 año de venetoclax y obinutuzumab. Los pacientes se asignaron al azar a recibir otro año de venetoclax, con independencia del estado de ERM, u otro año de venetoclax solo si eran positivos para ERM. Al cabo de una mediana de seguimiento de 35,2 meses, las tasas de ERM fueron idénticas en ambos grupos aleatorizados, lo que indica que este abordaje no mejora la eficacia, aunque se notificó un aumento de los eventos adversos con un año adicional de venetoclax.[47]
Antes de usar la ERM fuera del entorno de un ensayo clínico, se necesita evidencia concluyente que indique que la ERM es un biomarcador predictivo que puede guiar la toma de decisiones en la práctica clínica.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Byrd JC, Furman RR, Coutre SE, et al.: Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369 (1): 32-42, 2013.
O'Brien S, Furman RR, Coutre S, et al.: Single-agent ibrutinib in treatment-naïve and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood 131 (17): 1910-1919, 2018.
O'Brien S, Jones JA, Coutre SE, et al.: Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol 17 (10): 1409-1418, 2016.
Stilgenbauer S, Eichhorst B, Schetelig J, et al.: Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 17 (6): 768-78, 2016.
Stilgenbauer S, Eichhorst B, Schetelig J, et al.: Venetoclax for Patients With Chronic Lymphocytic Leukemia With 17p Deletion: Results From the Full Population of a Phase II Pivotal Trial. J Clin Oncol 36 (19): 1973-1980, 2018.
Brown JR, Eichhorst B, Hillmen P, et al.: Zanubrutinib or Ibrutinib in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 388 (4): 319-332, 2023.
Byrd JC, Hillmen P, Ghia P, et al.: Acalabrutinib Versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia: Results of the First Randomized Phase III Trial. J Clin Oncol 39 (31): 3441-3452, 2021.
Seymour JF, Byrd JC, Ghia P, et al.: Detailed safety profile of acalabrutinib vs ibrutinib in previously treated chronic lymphocytic leukemia in the ELEVATE-RR trial. Blood 142 (8): 687-699, 2023.
Bhat SA, Gambril J, Azali L, et al.: Ventricular arrhythmias and sudden death events following acalabrutinib initiation. Blood 140 (20): 2142-2145, 2022.
Woyach JA, Ruppert AS, Heerema NA, et al.: Ibrutinib Regimens versus Chemoimmunotherapy in Older Patients with Untreated CLL. N Engl J Med 379 (26): 2517-2528, 2018.
Burger JA, Sivina M, Jain N, et al.: Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood 133 (10): 1011-1019, 2019.
Shanafelt TD, Wang XV, Kay NE, et al.: Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med 381 (5): 432-443, 2019.
Shanafelt TD, Wang XV, Hanson CA, et al.: Long-term outcomes for ibrutinib-rituximab and chemoimmunotherapy in CLL: updated results of the E1912 trial. Blood 140 (2): 112-120, 2022.
Shanafelt TD, Wang V, Kay NE, et al.: Ibrutinib and rituximab provides superior clinical outcome compared to FCR in younger patients with chronic lymphocytic leukemia (CLL): extended follow-up from the E1912 trial. [Abstract] Blood 134 (Suppl 1): A-33, 2019.
Wang XV, Hanson CA, Tschumper RC, et al.: Measurable residual disease does not preclude prolonged progression-free survival in CLL treated with ibrutinib. Blood 138 (26): 2810-2827, 2021.
Tam CS, Brown JR, Kahl BS, et al.: Zanubrutinib versus bendamustine and rituximab in untreated chronic lymphocytic leukaemia and small lymphocytic lymphoma (SEQUOIA): a randomised, controlled, phase 3 trial. Lancet Oncol 23 (8): 1031-1043, 2022.
Sharman JP, Egyed M, Jurczak W, et al.: Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet 395 (10232): 1278-1291, 2020.
Fischer K, Al-Sawaf O, Bahlo J, et al.: Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N Engl J Med 380 (23): 2225-2236, 2019.
Al-Sawaf O, Zhang C, Tandon M, et al.: Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow-up results from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 21 (9): 1188-1200, 2020.
Al-Sawaf O, Zhang C, Lu T, et al.: Minimal Residual Disease Dynamics after Venetoclax-Obinutuzumab Treatment: Extended Off-Treatment Follow-up From the Randomized CLL14 Study. J Clin Oncol 39 (36): 4049-4060, 2021.
Seymour JF, Kipps TJ, Eichhorst B, et al.: Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N Engl J Med 378 (12): 1107-1120, 2018.
Kater AP, Seymour JF, Hillmen P, et al.: Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study. J Clin Oncol 37 (4): 269-277, 2019.
Seymour JF, Kipps TJ, Eichhorst BF, et al.: Four-year analysis of Murano study confirms sustained benefit of time-limited venetoclax-rituximab (VenR) in relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL). [Abstract] Blood 134 (Suppl 1): A-355, 2019.
Seymour JF, Kipps TJ, Eichhorst BF, et al.: Enduring undetectable MRD and updated outcomes in relapsed/refractory CLL after fixed-duration venetoclax-rituximab. Blood 140 (8): 839-850, 2022.
Eichhorst B, Fink AM, Bahlo J, et al.: First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 17 (7): 928-942, 2016.
Thompson PA, Tam CS, O'Brien SM, et al.: Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 127 (3): 303-9, 2016.
Fischer K, Bahlo J, Fink AM, et al.: Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 127 (2): 208-15, 2016.
Rossi D, Terzi-di-Bergamo L, De Paoli L, et al.: Molecular prediction of durable remission after first-line fludarabine-cyclophosphamide-rituximab in chronic lymphocytic leukemia. Blood 126 (16): 1921-4, 2015.
Munir T, Cairns DA, Bloor A, et al.: Chronic Lymphocytic Leukemia Therapy Guided by Measurable Residual Disease. N Engl J Med 390 (4): 326-337, 2024.
Niemann CU, Munir T, Moreno C, et al.: Fixed-duration ibrutinib-venetoclax versus chlorambucil-obinutuzumab in previously untreated chronic lymphocytic leukaemia (GLOW): 4-year follow-up from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol 24 (12): 1423-1433, 2023.
Wierda WG, Allan JN, Siddiqi T, et al.: Ibrutinib Plus Venetoclax for First-Line Treatment of Chronic Lymphocytic Leukemia: Primary Analysis Results From the Minimal Residual Disease Cohort of the Randomized Phase II CAPTIVATE Study. J Clin Oncol 39 (34): 3853-3865, 2021.
Hillmen P, Rawstron AC, Brock K, et al.: Ibrutinib Plus Venetoclax in Relapsed/Refractory Chronic Lymphocytic Leukemia: The CLARITY Study. J Clin Oncol 37 (30): 2722-2729, 2019.
Tam CS, Allan JN, Siddiqi T, et al.: Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood 139 (22): 3278-3289, 2022.
Huber H, Tausch E, Schneider C, et al.: Final analysis of the CLL2-GIVe trial: obinutuzumab, ibrutinib, and venetoclax for untreated CLL with del(17p)/TP53mut. Blood 142 (11): 961-972, 2023.
Hillmen P, Eichhorst B, Brown JR, et al.: Zanubrutinib Versus Ibrutinib in Relapsed/Refractory Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma: Interim Analysis of a Randomized Phase III Trial. J Clin Oncol 41 (5): 1035-1045, 2023.
Shadman M, Flinn IW, Levy MY, et al.: Zanubrutinib in patients with previously treated B-cell malignancies intolerant of previous Bruton tyrosine kinase inhibitors in the USA: a phase 2, open-label, single-arm study. Lancet Haematol 10 (1): e35-e45, 2023.
Ma S, Seymour JF, Brander DM, et al.: Efficacy of venetoclax plus rituximab for relapsed CLL: 5-year follow-up of continuous or limited- duration therapy. Blood 138 (10): 836-846, 2021.
Allan JN, Shanafelt T, Wiestner A, et al.: Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukaemia in patients with TP53 aberrations: a pooled analysis from four clinical trials. Br J Haematol 196 (4): 947-953, 2022.
Jain N, Keating M, Thompson P, et al.: Ibrutinib and Venetoclax for First-Line Treatment of CLL. N Engl J Med 380 (22): 2095-2103, 2019.
Rogers KA, Huang Y, Ruppert AS, et al.: Phase II Study of Combination Obinutuzumab, Ibrutinib, and Venetoclax in Treatment-Naïve and Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol 38 (31): 3626-3637, 2020.
Soumerai JD, Mato AR, Dogan A, et al.: Zanubrutinib, obinutuzumab, and venetoclax with minimal residual disease-driven discontinuation in previously untreated patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: a multicentre, single-arm, phase 2 trial. Lancet Haematol 8 (12): e879-e890, 2021.
Huber H, Edenhofer S, von Tresckow J, et al.: Obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) frontline treatment for high-risk chronic lymphocytic leukemia. Blood 139 (9): 1318-1329, 2022.
Ahn IE, Tian X, Ipe D, et al.: Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia Treated With Ibrutinib: Development and Validation of a Four-Factor Prognostic Model. J Clin Oncol 39 (6): 576-585, 2021.
Kovacs G, Robrecht S, Fink AM, et al.: Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J Clin Oncol 34 (31): 3758-3765, 2016.
Thompson PA, Srivastava J, Peterson C, et al.: Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood 134 (22): 1951-1959, 2019.
Kater AP, Levin MD, Dubois J, et al.: Minimal residual disease-guided stop and start of venetoclax plus ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia (HOVON141/VISION): primary analysis of an open-label, randomised, phase 2 trial. Lancet Oncol 23 (6): 818-828, 2022.
Kersting S, Dubois J, Nasserinejad K, et al.: Venetoclax consolidation after fixed-duration venetoclax plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (HOVON 139/GiVe): primary endpoint analysis of a multicentre, open-label, randomised, parallel-group, phase 2 trial. Lancet Haematol 9 (3): e190-e199, 2022.
Tratamiento de la leucemia linfocítica crónica recidivante o resistente al tratamiento
Opciones de tratamiento para la leucemia linfocítica crónica recidivante o resistente al tratamiento
Es posible administrar de nuevo y en forma secuencial los mismos regímenes que se usan para la terapia de primera línea en los pacientes con leucemia linfocítica crónica (LLC). Estos regímenes se describen en detalle en la sección sobre el tratamiento de primera línea. Para obtener más información, consultar la sección Tratamiento de la leucemia linfocítica crónica sintomática o progresiva.
Inhibidores de la tirosina–cinasa de Bruton (BTK) (acalabrutinib, zanubrutinib o ibrutinib).
Venetoclax con uso inicial de obinutuzumab o rituximab.
Bendamustina y rituximab (BR).
Fludarabina, ciclofosfamida y rituximab (FCR).
Inhibidor de la BTK (ibrutinib o acalabrutinib) y venetoclax.
R-CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina y prednisona) (solo si hay sospecha clínica de síndrome de Richter con progresión histológica).
En el entorno de una recaída, el venetoclax exhibió similaridad en la eficacia y la inocuidad, incluso después del tratamiento previo con ibrutinib o idelalisib (inhibidor de la fosfatidilinositol 3 cinasa [PI3K] δ).[1,2]
Asimismo, en un ensayo publicado como resumen, el ibrutinib y el acalabrutinib exhibieron similaridad en la eficacia y la inocuidad después del antecedente de terapia con venetoclax.[3] La administración en secuencia de estos nuevos fármacos demostró eficacia en el entorno de recaída o resistencia al tratamiento.[4,5]
Inhibidores de la tirosina–cinasa de Bruton no covalentes
A diferencia de otros inhibidores de la tirosina–cinasa de Bruton (BTK), como ibrutinib, acalabrutinib y zanubrutinib, pirtobrutinib se une a la BTK mediante un enlace no covalente.[6]
En un estudio prospectivo de fase I/II, 317 pacientes con LLC o leucemia linfocítica pequeña recibieron pirtobrutinib. Un total de 247 de estos pacientes habían recibido tratamiento previo con un inhibidor de la BTK d
Al cabo de una mediana de seguimiento de 19,4 meses, la mediana de supervivencia sin progresión (SSP) fue de 19,6 meses (intervalo de confianza [IC] 95 %, 16,9–22,1) para todos los pacientes y de 16,8 meses (IC 95 %, 13,2–18,7) para el subgrupo de pacientes con resistencia a los inhibidores de la BTK, intolerancia o variantes de BTK C481 (variante de resistencia a los fármacos covalentes).[7][Nivel de evidencia C3]
Terapia de células T con receptor de antígeno quimérico
Las células T autógenas se pueden modificar mediante vectores virales para incorporar un receptor específico del antígeno de células B CD19 y, luego, se administran mediante infusión a pacientes que recibieron tratamiento previo.[8] La respuesta sorprendente, que duró 6 meses, dio lugar a ensayos más grandes a partir de este concepto. En ensayos clínicos en curso se está probando este concepto de usar células T dirigidas a CD19 mediante la administración de células T con CAR genotecnológicos.[9,10,11]
Inhibidores de PI3K
El idelalisib es un fármaco de uso oral que actúa como inhibidor de la isoforma δ de la PI3K, que participa en la secuencia en cadena de señalización del receptor de células B. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) retiró la indicación del idelalisib debido a su toxicidad y ya no está disponible. El duvelisib es un fármaco de uso oral que actúa como inhibidor doble de las isoformas δ y γ de PI3K.[12]
En un ensayo prospectivo, 319 pacientes con LLC o linfoma linfocítico pequeño en recaída o resistentes al tratamiento, se asignaron al azar a recibir duvelisib versus ofatumumab.[13]
Al cabo de una mediana de seguimiento de 22,4 meses, la mediana de SSP en los pacientes que recibieron duvelisib fue de 13,3 meses, significativamente más prolongada que en los pacientes que recibieron ofatumumab, que fue de 9,9 meses (cociente de riesgos instantáneos [CRI], 0,52; P < 0,0001).[13][Nivel de evidencia B1]
Los efectos secundarios graves incluyen infecciones, neumonitis, diarrea o colitis, dermatitis, neutropenia, exantema, fatiga, fiebre, náuseas, anemia y transaminitis.
La FDA ha aprobado el duvelisib para su uso como tratamiento de tercera línea (o superior). Sin embargo, los datos de seguimiento presentados a la FDA mostraron, al cabo de una mediana de seguimiento de 63 meses, una mediana de SG de 52,3 meses (IC 95 %, 41,8–68,0) en el grupo de duvelisib y de 63,3 meses (IC 95 %, 41,2– no estimable) en el grupo de ofatumumab.[14]
Lenalidomida (con rituximab o sin este)
La lenalidomida es un inmunomodulador oral con tasas de respuesta superiores al 50 % cuando se usa con rituximab o sin este, en pacientes con enfermedad tratada o sin tratar.[15,16,17,18,19,20,21][Nivel de evidencia C3] Al usar este fármaco, se recomiendan abordajes de dosis bajas prolongados y poner atención en la prevención del síndrome de lisis tumoral.[15,22] En los pacientes con LLC todavía no se ha definido la terapia combinada y los efectos tóxicos a largo plazo cuando se usa la lenalidomida (como un aumento de la mielodisplasia, que se observa en los pacientes de mieloma).
Trasplante de médula ósea o de células madre periféricas
En un ensayo prospectivo aleatorizado, 241 menores de 65 años que no recibieron tratamiento previo y que tenían enfermedad en estadio avanzado recibieron una terapia de inducción con un régimen a base de ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) seguido de fludarabina.[23] Los pacientes que presentaron una respuesta completa (105 pacientes) se asignaron al azar a recibir un trasplante de células madre (TCM) autógeno u observación, mientras que los otros 136 pacientes se asignaron al azar a recibir una reinducción con dexametasona, dosis altas de citarabina y cisplatino, seguida de TCM autógeno o fludarabina con ciclofosfamida (FC). Si bien la supervivencia sin complicaciones (SSC) a 3 años favoreció el uso de TCM autógeno en los pacientes con respuesta completa, no hubo diferencia de SG en ninguna de las comparaciones aleatorizadas.[23][Nivel de evidencia B1] El trasplante autógeno de médula ósea o de células madre se emplea con poca frecuencia para los pacientes con LLC en recaída.
Los pacientes con factores de pronóstico adverso tienen muchas probabilidades de morir por la LLC. Estos pacientes son aptos para participar en ensayos clínicos en los que se emplean dosis altas de quimioterapia e inmunoterapia con TCM de sangre periférica alogénico mielosupresor y no mielosupresor .[24,25,26,27,28,29] Aunque la mayoría de los pacientes que logran una remisión completa luego de un TCM autógeno recaen con el tiempo; la meseta de supervivencia del TCM alogénico indica un efecto adicional de injerto contra leucemia.[29] En una serie (NCT00281983) de 90 pacientes con LLC en recaída o resistente al tratamiento que recibieron un TCM alogénico, se notificó una tasa de SG a 6 años del 58 % y una tasa de SSC a 6 años del 38 %. Este grupo incluyó pacientes con los peores factores pronósticos (como una variante patogénica del gen TP53).[30][Nivel de evidencia C2]
Ofatumumab
El ofatumumab es un anticuerpo monoclonal humanizado anti-CD20.
Evidencia (ofatumumab solo y en combinación con clorambucilo):
En un ensayo prospectivo participaron 474 pacientes que recibieron tratamiento previo que alcanzaron una remisión parcial o completa con quimioterapia de segunda o tercera línea. Los pacientes se asignaron al azar a recibir 2 años de terapia de mantenimiento con ofatumumab versus observación.[31]
Después de una mediana de seguimiento de 19 meses, la mediana de SSP favoreció al grupo de mantenimiento con ofatumumab a los 29,4 meses versus 15,2 meses (CRI, 0,50; IC 95 %, 0,38–0,66, P < 0,0001). No hubo ninguna diferencia en la SG.[31][Nivel de evidencia B1]
En un ensayo prospectivo aleatorizado con 447 pacientes sin tratamiento previo se comparó ofatumumab y clorambucilo con clorambucilo solo.[32]
Después de una mediana de seguimiento de 2 años, la mediana de SSP favoreció al grupo de ofatumumab y clorambucilo a los 22,4 meses versus 13,1 meses (CRI, 0,57; IC 95 %, 0,45–0,72; P = 0,0001). No hubo ninguna diferencia en la SG.[32][Nivel de evidencia B1]
Radioterapia dirigida al campo comprometido
Es posible administrar dosis relativamente bajas de radioterapia para las linfadenopatías que causan problemas por el tamaño o la invasión de órganos adyacentes. Algunas veces la radioterapia dirigida a un área ganglionar o al bazo produce un efecto abscopal (es decir, achicamiento de ganglios linfáticos en sitios no tratados).
El alemtuzumab, un anticuerpo monoclonal dirigido a CD52, exhibe actividad en el entorno de la enfermedad resistente a la quimioterapia, o en pacientes no tratados de riesgo alto que tienen del(17p) o variantes patogénicas de TP53.[33,34,35] Cuando se administra en monoterapia, la vía de administración subcutánea se prefiere a la vía intravenosa porque su eficacia es similar, pero con menos efectos adversos, por ejemplo, menos reacciones alérgicas graves según se observó en algunos informes de estudios no aleatorizados.[35,36,37,38,39]
Usado en un régimen combinado, el alemtuzumab subcutáneo y la fludarabina (con ciclofosfamida o sin esta) o el alemtuzumab intravenoso acompañado de alquilantes, produjo demasiada toxicidad infecciosa y mortalidad, sin mejoría compensatoria en la eficacia en 3 ensayos de fase II y en un ensayo aleatorizado.[40,41,42][Nivel de evidencia C3]; [43][Nivel de evidencia B1]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Jones JA, Mato AR, Wierda WG, et al.: Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol 19 (1): 65-75, 2018.
Coutre S, Choi M, Furman RR, et al.: Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood 131 (15): 1704-1711, 2018.
Kater AP, Wu JQ, Kipps T, et al.: Venetoclax Plus Rituximab in Relapsed Chronic Lymphocytic Leukemia: 4-Year Results and Evaluation of Impact of Genomic Complexity and Gene Mutations From the MURANO Phase III Study. J Clin Oncol 38 (34): 4042-4054, 2020.
Mato AR, Hill BT, Lamanna N, et al.: Optimal sequencing of ibrutinib, idelalisib, and venetoclax in chronic lymphocytic leukemia: results from a multicenter study of 683 patients. Ann Oncol 28 (5): 1050-1056, 2017.
Kater AP, Arslan Ö, Demirkan F, et al.: Activity of venetoclax in patients with relapsed or refractory chronic lymphocytic leukaemia: analysis of the VENICE-1 multicentre, open-label, single-arm, phase 3b trial. Lancet Oncol 25 (4): 463-473, 2024.
Thompson PA, Tam CS: Pirtobrutinib: a new hope for patients with BTK inhibitor-refractory lymphoproliferative disorders. Blood 141 (26): 3137-3142, 2023.
Mato AR, Woyach JA, Brown JR, et al.: Pirtobrutinib after a Covalent BTK Inhibitor in Chronic Lymphocytic Leukemia. N Engl J Med 389 (1): 33-44, 2023.
Porter DL, Levine BL, Kalos M, et al.: Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365 (8): 725-33, 2011.
Turtle CJ, Hay KA, Hanafi LA, et al.: Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated With CD19-Specific Chimeric Antigen Receptor-Modified T Cells After Failure of Ibrutinib. J Clin Oncol 35 (26): 3010-3020, 2017.
Siddiqi T, Soumerai JD, Dorritie KA: Rapid undetectable MRD (uMRD) responses in patients with relapsed/refractory (R/R) chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) treated with lisocabtagene maraleucel (liso-cel), a CD19-directed CAR T cell product: updated results from Transcend CLL 004, a phase 1/2 study including patients with high-risk disease previously treated with ibrutinib. [Abstract] Blood 134 (Suppl 1): 503, 2019.
Frey NV, Gill S, Hexner EO, et al.: Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. J Clin Oncol 38 (25): 2862-2871, 2020.
Patel K, Danilov AV, Pagel JM: Duvelisib for CLL/SLL and follicular non-Hodgkin lymphoma. Blood 134 (19): 1573-1577, 2019.
Flinn IW, Hillmen P, Montillo M, et al.: The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood 132 (23): 2446-2455, 2018.
U.S. Food and Drug Administration: FDA warns about possible increased risk of death and serious side effects with cancer drug Copiktra (duvelisib). Silver Spring, Md: U.S. Food and Drug Administration, 2022. Available online. Last accessed April 3, 2025.
Chen CI, Bergsagel PL, Paul H, et al.: Single-agent lenalidomide in the treatment of previously untreated chronic lymphocytic leukemia. J Clin Oncol 29 (9): 1175-81, 2011.
Chanan-Khan A, Miller KC, Musial L, et al.: Clinical efficacy of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia: results of a phase II study. J Clin Oncol 24 (34): 5343-9, 2006.
Ferrajoli A, Lee BN, Schlette EJ, et al.: Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood 111 (11): 5291-7, 2008.
Strati P, Keating MJ, Wierda WG, et al.: Lenalidomide induces long-lasting responses in elderly patients with chronic lymphocytic leukemia. Blood 122 (5): 734-7, 2013.
Wendtner CM, Hillmen P, Mahadevan D, et al.: Final results of a multicenter phase 1 study of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia. Leuk Lymphoma 53 (3): 417-23, 2012.
Badoux XC, Keating MJ, Wen S, et al.: Phase II study of lenalidomide and rituximab as salvage therapy for patients with relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol 31 (5): 584-91, 2013.
Takahashi K, Hu B, Wang F, et al.: Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 131 (16): 1820-1832, 2018.
Sutton L, Chevret S, Tournilhac O, et al.: Autologous stem cell transplantation as a first-line treatment strategy for chronic lymphocytic leukemia: a multicenter, randomized, controlled trial from the SFGM-TC and GFLLC. Blood 117 (23): 6109-19, 2011.
Toze CL, Dalal CB, Nevill TJ, et al.: Allogeneic haematopoietic stem cell transplantation for chronic lymphocytic leukaemia: outcome in a 20-year cohort. Br J Haematol 158 (2): 174-85, 2012.
Khouri IF, Saliba RM, Admirand J, et al.: Graft-versus-leukaemia effect after non-myeloablative haematopoietic transplantation can overcome the unfavourable expression of ZAP-70 in refractory chronic lymphocytic leukaemia. Br J Haematol 137 (4): 355-63, 2007.
Sorror ML, Storer BE, Sandmaier BM, et al.: Five-year follow-up of patients with advanced chronic lymphocytic leukemia treated with allogeneic hematopoietic cell transplantation after nonmyeloablative conditioning. J Clin Oncol 26 (30): 4912-20, 2008.
Schetelig J, van Biezen A, Brand R, et al.: Allogeneic hematopoietic stem-cell transplantation for chronic lymphocytic leukemia with 17p deletion: a retrospective European Group for Blood and Marrow Transplantation analysis. J Clin Oncol 26 (31): 5094-100, 2008.
Malhotra P, Hogan WJ, Litzow MR, et al.: Long-term outcome of allogeneic stem cell transplantation in chronic lymphocytic leukemia: analysis after a minimum follow-up of 5 years. Leuk Lymphoma 49 (9): 1724-30, 2008.
Dreger P, Döhner H, Ritgen M, et al.: Allogeneic stem cell transplantation provides durable disease control in poor-risk chronic lymphocytic leukemia: long-term clinical and MRD results of the German CLL Study Group CLL3X trial. Blood 116 (14): 2438-47, 2010.
Dreger P, Schnaiter A, Zenz T, et al.: TP53, SF3B1, and NOTCH1 mutations and outcome of allotransplantation for chronic lymphocytic leukemia: six-year follow-up of the GCLLSG CLL3X trial. Blood 121 (16): 3284-8, 2013.
van Oers MH, Kuliczkowski K, Smolej L, et al.: Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol 16 (13): 1370-9, 2015.
Hillmen P, Robak T, Janssens A, et al.: Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 385 (9980): 1873-83, 2015.
Moreton P, Kennedy B, Lucas G, et al.: Eradication of minimal residual disease in B-cell chronic lymphocytic leukemia after alemtuzumab therapy is associated with prolonged survival. J Clin Oncol 23 (13): 2971-9, 2005.
Parikh SA, Keating MJ, O'Brien S, et al.: Frontline chemoimmunotherapy with fludarabine, cyclophosphamide, alemtuzumab, and rituximab for high-risk chronic lymphocytic leukemia. Blood 118 (8): 2062-8, 2011.
Pettitt AR, Jackson R, Carruthers S, et al.: Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. J Clin Oncol 30 (14): 1647-55, 2012.
Stilgenbauer S, Zenz T, Winkler D, et al.: Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 27 (24): 3994-4001, 2009.
Cortelezzi A, Pasquini MC, Gardellini A, et al.: Low-dose subcutaneous alemtuzumab in refractory chronic lymphocytic leukaemia (CLL): results of a prospective, single-arm multicentre study. Leukemia 23 (11): 2027-33, 2009.
Osterborg A, Foà R, Bezares RF, et al.: Management guidelines for the use of alemtuzumab in chronic lymphocytic leukemia. Leukemia 23 (11): 1980-8, 2009.
Gritti G, Reda G, Maura F, et al.: Low dose alemtuzumab in patients with fludarabine-refractory chronic lymphocytic leukemia. Leuk Lymphoma 53 (3): 424-9, 2012.
Lin TS, Donohue KA, Byrd JC, et al.: Consolidation therapy with subcutaneous alemtuzumab after fludarabine and rituximab induction therapy for previously untreated chronic lymphocytic leukemia: final analysis of CALGB 10101. J Clin Oncol 28 (29): 4500-6, 2010.
Badoux XC, Keating MJ, Wang X, et al.: Cyclophosphamide, fludarabine, alemtuzumab, and rituximab as salvage therapy for heavily pretreated patients with chronic lymphocytic leukemia. Blood 118 (8): 2085-93, 2011.
Lepretre S, Aurran T, Mahé B, et al.: Excess mortality after treatment with fludarabine and cyclophosphamide in combination with alemtuzumab in previously untreated patients with chronic lymphocytic leukemia in a randomized phase 3 trial. Blood 119 (22): 5104-10, 2012.
Geisler CH, van T' Veer MB, Jurlander J, et al.: Frontline low-dose alemtuzumab with fludarabine and cyclophosphamide prolongs progression-free survival in high-risk CLL. Blood 123 (21): 3255-62, 2014.
Referencias bibliográficas clave para la leucemia linfocítica crónica
Los miembros del Consejo editorial del PDQ sobre el tratamiento para adultos seleccionaron estas referencias bibliográficas por su importancia en el entorno del tratamiento de la leucemia linfocítica crónica (LLC). Esta lista se proporciona para informar a los interesados sobre estudios importantes que han ayudado a conformar la comprensión actual sobre las opciones de tratamiento para la leucemia linfocítica crónica. A continuación, se indican las secciones del resumen en las que se citaron las referencias.
Fischer K, Al-Sawaf O, Bahlo J, et al.: Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N Engl J Med 380 (23): 2225-2236, 2019. [PUBMED Abstract]
Shanafelt TD, Wang XV, Kay NE, et al.: Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N Engl J Med 381 (5): 432-443, 2019. [PUBMED Abstract]
Shanafelt TD, Wang V, Kay NE, et al.: Ibrutinib and rituximab provides superior clinical outcome compared to FCR in younger patients with chronic lymphocytic leukemia (CLL): extended follow-up from the E1912 trial. [Abstract] Blood 134 (Suppl 1): A-33, 2019.
Al-Sawaf O, Zhang C, Tandon M, et al.: Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow-up results from a multicentre, open-label, randomised, phase 3 trial. Lancet Oncology 21 (9): 1188-1200, 2020. [PUBMED Abstract]
Actualizaciones más recientes a este resumen (04 / 23 / 2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan en la medida en que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos a este resumen a partir de la fecha arriba indicada.
Se actualizaron las estadísticas con el número estimado de casos nuevos y defunciones para 2025 (se citó a la American Cancer Society como referencia 1).
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento de la leucemia linfocítica crónica. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento para adultos, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
El revisor principal del sumario sobre Tratamiento de la leucemia linfocítica crónica es:
Eric J. Seifter, MD (Johns Hopkins University)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento para adultos emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.