Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre la leucemia linfoblástica aguda
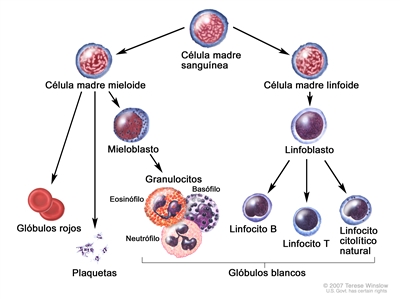
La leucemia linfoblástica aguda (también llamada leucemia linfocítica aguda o LLA) es un tipo de leucemia muy maligna que se caracteriza por la presencia de demasiados linfoblastos o linfocitos en la médula ósea y la sangre periférica. Es posible que se disemine a los ganglios linfáticos, el bazo, el hígado, el sistema nervioso central (SNC), los testículos u otros órganos. Sin tratamiento, la LLA suele evolucionar de forma rápida.
Entre los signos y síntomas de LLA se encuentran los siguientes:
Debilidad o fatiga.
Fiebre o sudoración nocturna.
Equimosis o hemorragias que se producen con facilidad (es decir, hemorragia gingival, hematomas cutáneos o petequias [hemorragias puntiformes bajo la piel]).
Dificultad respiratoria.
Pérdida de peso inesperada o anorexia.
Dolor óseo o articular.
Adenopatías, en especial, en el cuello, las axilas, o la ingle, por lo general, indoloras.
Hinchazón, distensión o incomodidad en el abdomen.
Infecciones frecuentes.
La LLA se presenta tanto en niños como en adultos. Es el tipo de cáncer más común en los niños y el tratamiento produce una buena probabilidad de curación. Para los adultos, el pronóstico no es tan optimista. En este resumen se analiza la LLA en personas adultas. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda infantil.
Incidencia y mortalidad
Número estimado de casos nuevos y defunciones por LLA en los Estados Unidos para 2025:[1]
Casos nuevos: 6100.
Defunciones: 1400.
Características anatómicas
Se piensa que la LLA surge de la transformación maligna de las células progenitoras B o T.[2] Se observa con mayor frecuencia en los niños, pero es posible que se presente a cualquier edad. La enfermedad se caracteriza por la acumulación de linfoblastos en la médula o en varios sitios extramedulares, con frecuencia acompañada de supresión de la hematopoyesis normal. Las células leucémicas linfoblásticas B y T expresan antígenos de superficie muy similares a los característicos del desarrollo de sus respectivos linajes. Por lo general, las LLA de células precursoras B expresan CD10, CD19 y CD34 en su superficie junto con la desoxinucleotidil–transferasa terminal (TdT) nuclear, mientras que la LLA de células precursoras T expresan CD2, CD3, CD7, CD34 y TdT en la mayoría de los casos.
Características moleculares
Es posible que algunos pacientes que presentan leucemia aguda tengan una anomalía citogenética que no se puede distinguir de manera citogenética del cromosoma Filadelfia (Ph).[3] El Ph se presenta solo en el 1 % al 2 % de los pacientes con leucemia mieloide aguda (LMA), pero se presenta en cerca del 20 % de los adultos y en un porcentaje pequeño de niños con LLA.[4] En la mayoría de los niños, y en más de la mitad de los adultos con LLA positiva para el cromosoma Ph, la anomalía molecular es diferente de la que se presenta en la leucemia mieloide crónica (LMC) positiva para Ph.
Muchos pacientes con constancia molecular del gen de fusión BCR::ABL1, que caracteriza el Ph, no exhiben ningún cromosoma anormal en el análisis citogenético. Es posible que el gen de fusión BCR::ABL1 solo se detecte mediante hibridación fluorescente in situ (FISH) o reacción en cadena de la polimerasa con retrotranscripción (RCP-RT), debido a que muchos pacientes tienen una proteína de fusión diferente de la que se encuentra en la LMC (p190 vs. p210). Estos exámenes se deberán llevar a cabo cuando sea posible en pacientes con LLA, en especial aquellos con enfermedad de linaje de células B.
La LLA L3 está relacionada con una variedad de translocaciones que incluye el protooncogén MYC y el locus del gen de inmunoglobulina t(2;8), t(8;12) y t(8;22).
Diagnóstico
Los pacientes con LLA tal vez presenten varias alteraciones hematológicas, que oscilan de pancitopenia a hiperleucocitosis. Además de los antecedentes y del examen físico, la evaluación diagnóstica inicial debe incluir los siguientes procedimientos:
Fibrinógenos y pruebas de coagulación, como un examen de detección de coagulación intravascular diseminada.
Evaluación cuidadosa en busca de indicios de infección activa.
Con frecuencia, se lleva a cabo de forma rutinaria una biopsia de la médula ósea y un aspirado, incluso en las LLA de células T, para determinar la extensión del compromiso medular. Se debe realizar un estudio citogenético convencional de las células malignas, ya que la detección de Ph t(9;22), los reordenamientos génicos de MYC (en la leucemia de Burkitt) y los reordenamientos génicos de MLL añaden información pronóstica importante. La citometría de flujo se debe llevar a cabo para caracterizar los antígenos que definen la expresión de linaje y determinar los subtipos específicos de la LLA. Además, para la enfermedad de células B, las células malignas se deben analizar por medio de RCP-RT y FISH para detectar el gen de fusión BCR::ABL1. Este último punto es de suma importancia, ya que un diagnóstico a tiempo de la LLA Ph cambiaría el abordaje terapéutico de manera significativa.
Es posible que confunda el diagnóstico con el de la leucemia mielocítica aguda (LMA), la leucemia de células pilosas y el linfoma maligno. Es de suma importancia efectuar un diagnóstico adecuado debido a las diferencias en el pronóstico y el tratamiento entre la LLA y la LMA. El análisis inmunofenotípico es esencial porque las leucemias que no expresan mieloperoxidasa son las LMA M0, LMA M7 y LLA.
El examen de los aspirados de médula ósea o de las piezas de biopsia lo debe efectuar un oncólogo, un hematólogo, un hematopatólogo o un patólogo general experto y capaz de interpretar piezas convencionales y, en especial, piezas teñidas.
Pronóstico y supervivencia
Entre los factores relacionados con el pronóstico de pacientes con LLA se encuentran los siguientes:
Edad: la edad, un factor significativo en la LLA y en la LMA infantiles, quizás sea un factor pronóstico importante en la LLA en adultos. En un estudio, el pronóstico fue mejor en general en pacientes menores de 25 años; en otro estudio, se observó un pronóstico mejor para los pacientes menores de 35 años. Es posible que estos hallazgos se relacionen en cierta forma con una mayor incidencia del Ph en pacientes de más edad con LLA, un subgrupo relacionado con un pronóstico precario.[5,6]
Compromiso del SNC: al igual que en la LLA infantil, los pacientes adultos con LLA tienen riesgo de presentar complicaciones del sistema nervioso central (SNC) durante el curso de la enfermedad. En especial, esto es cierto en aquellos pacientes con un tipo morfolófico L3 (Burkitt).[7] Tanto el tratamiento como el pronóstico se ven afectados por esta complicación.
Morfología celular: los pacientes con un tipo morfológico L3 muestran resultados mejores, según consta en el estudio Cancer and Leukemia Group B (CLB-9251 [NCT00002494]), que ya se completó, cuando se tratan de acuerdo con algoritmos de tratamiento específicos.[8,9] En este estudio se descubrió que es posible curar la leucemia L3 mediante regímenes quimioterapéuticos intensivos de ciclos rápidos tipo linfoma.[8,10,11]
Anomalías cromosómicas: se han descrito anomalías cromosómicas, como aneuploidía y translocaciones, que quizás se correlacionen con el pronóstico.[12] En particular, los pacientes con LLA positiva para Ph t(9;22) tienen un pronóstico precario y representan más del 30 % de los casos en adultos. Los pacientes con leucemia y un gen de fusión BCR::ABL1 que no muestran el Ph clásico presentan un pronóstico precario similar al de la LLA positiva para Ph. Los pacientes con LLA positiva para Ph son difíciles de curar con quimioterapia, aunque en la actualidad se informa con frecuencia sobre casos de supervivencia a largo plazo cuando dichos pacientes se tratan con combinaciones de quimioterapia e inhibidores de tirosina–cinasas de BCR::ABL1.
Otras dos anomalías cromosómicas relacionadas con un pronóstico precario son t(4;11), que se caracteriza por reordenamientos del gen MLL y que también es posible que se encuentre reordenado a pesar de otras características citogenéticas normales, y t(9;22). Además de t(4;11) y t(9;22), se ha informado que los pacientes con deleción del cromosoma 7 o trisomía 8 tienen menos probabilidades de supervivencia a 5 años cuando se les compara con pacientes que presentan cariotipo normal.[13] En un análisis multivariante, el cariotipo fue el factor pronóstico más importante de supervivencia sin enfermedad.[13][Nivel de evidencia C2]
Efectos tardíos del tratamiento de la leucemia linfoblástica aguda
En un seguimiento a largo plazo de 30 pacientes con LLA en remisión durante al menos 10 años se observaron 10 casos de neoplasias malignas secundarias. De 31 mujeres con supervivencia prolongada con LLA o LMA menores de 40 años, 26 continuaron con menstruación normal después de terminar el tratamiento. Entre los 36 hijos de estas sobrevivientes que nacieron vivos, se presentaron dos problemas congénitos.[14]
Referencias:
American Cancer Society: Cancer Facts and Figures 2025. American Cancer Society, 2025. Available online. Last accessed January 16, 2025.
Pui CH, Jeha S: New therapeutic strategies for the treatment of acute lymphoblastic leukaemia. Nat Rev Drug Discov 6 (2): 149-65, 2007.
Peterson LC, Bloomfield CD, Brunning RD: Blast crisis as an initial or terminal manifestation of chronic myeloid leukemia: a study of 28 patients. Am J Med 60(2): 209-220, 1976.
Secker-Walker LM, Cooke HM, Browett PJ, et al.: Variable Philadelphia breakpoints and potential lineage restriction of bcr rearrangement in acute lymphoblastic leukemia. Blood 72 (2): 784-91, 1988.
Gaynor J, Chapman D, Little C, et al.: A cause-specific hazard rate analysis of prognostic factors among 199 adults with acute lymphoblastic leukemia: the Memorial Hospital experience since 1969. J Clin Oncol 6 (6): 1014-30, 1988.
Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988.
Kantarjian HM, Walters RS, Smith TL, et al.: Identification of risk groups for development of central nervous system leukemia in adults with acute lymphocytic leukemia. Blood 72 (5): 1784-9, 1988.
Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001.
Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
Fenaux P, Lai JL, Miaux O, et al.: Burkitt cell acute leukaemia (L3 ALL) in adults: a report of 18 cases. Br J Haematol 71 (3): 371-6, 1989.
Reiter A, Schrappe M, Ludwig WD, et al.: Favorable outcome of B-cell acute lymphoblastic leukemia in childhood: a report of three consecutive studies of the BFM group. Blood 80 (10): 2471-8, 1992.
Chromosomal abnormalities and their clinical significance in acute lymphoblastic leukemia. Third International Workshop on Chromosomes in Leukemia. Cancer Res 43 (2): 868-73, 1983.
Wetzler M, Dodge RK, Mrózek K, et al.: Prospective karyotype analysis in adult acute lymphoblastic leukemia: the cancer and leukemia Group B experience. Blood 93 (11): 3983-93, 1999.
Micallef IN, Rohatiner AZ, Carter M, et al.: Long-term outcome of patients surviving for more than ten years following treatment for acute leukaemia. Br J Haematol 113 (2): 443-5, 2001.
Clasificación celular de la leucemia linfoblástica aguda
Las siguientes características celulares leucémicas son importantes:
Características morfológicas.
Características citogenéticas.
Marcadores inmunitarios de superficie celular y bioquímicos.
Característica citoquímicas.
En las personas adultas, el tipo morfológico L1 de la clasificación French-American-British (FAB) (linfoblastos de aspecto más maduro) está presente en menos del 50 % de los pacientes y predomina el tipo histológico L2 (más inmaduros y polimorfos).[1] La leucemia linfoblástica aguda (LLA) L3 (Burkitt) es mucho menos común que los otros dos subtipos FAB. Se caracteriza por blastocitos con vacuolas citoplasmáticas y expresión de inmunoglobulina de superficie, y con frecuencia, la médula ósea presenta una apariencia que se describe como cielo estrellado lo que se debe a la presencia de muchas células apoptósicas. La LLA L3 se relaciona con una variedad de translocaciones que incluye el protooncogén MYC y el locus del gen de inmunoglobulina t(2;8), t(8;12) y t(8;22).
Es posible que algunos pacientes que presentan leucemia aguda tengan una anomalía citogenética que no se puede distinguir morfológicamente del cromosoma Filadelfia (Ph).[2] El Ph se presenta solo en el 1 % al 2 % de los pacientes con leucemia mieloide aguda (LMA), pero se presenta en cerca del 20 % de los adultos y en un porcentaje pequeño de niños con LLA.[3] En la mayoría de los niños, y en más de la mitad de los adultos con LLA positiva para Ph, la anomalía molecular es diferente de la que se presenta en la leucemia mieloide crónica (LMC) positiva para Ph.
Muchos pacientes con constancia molecular del gen de fusión BCR::ABL1, que caracteriza el Ph, no exhiben ningún cromosoma anormal en el análisis citogenético. Es posible que el gen de fusión BCR::ABL1 solo sea detectable mediante electroforesis en gel de campo pulsado o reacción en cadena de la polimerasa con retrotranscripción, debido a que muchos pacientes tienen una proteína de fusión diferente de la que se encuentra en la LMC (p190 vs. p210).
Es posible dividir las células de LLA en diferentes subtipos mediante el uso de heteroantisueros y anticuerpos monoclonales (ver el Cuadro 1).[1,4,5,6]
Cuadro 1. Frecuencia de los subtipos celulares de la leucemia linfoblástica aguda
Subtipo celular
Frecuencia aproximada
Linaje de células B tempranas
80 %
Células T
10–15 %
Células B con inmunoglobulinas de superficie
<5 %
Cerca del 95 % de todos los tipos de LLA (excepto la de Burkitt que, por lo general, tiene un tipo morfológico L3 según la clasificación FAB) tienen una expresión elevada de desoxinucleotidil–transferasa terminal (TdT). Esta elevación es extremadamente útil en el diagnóstico; si las concentraciones de la enzima no son elevadas, entonces es posible sospechar que el diagnóstico sea LLA. Sin embargo, el 20 % de los casos de LMA expresan TdT; por lo tanto, su utilidad como marcador de linaje es limitada. Debido a que las leucemias de Burkitt se tratan de acuerdo con diferentes algoritmos de tratamiento, es importante identificar prospectivamente estos casos de forma específica por su tipo morfológico L3, ausencia de TdT y la expresión de la inmunoglobulina de superficie. Por lo general, los pacientes con leucemias de Burkitt tendrán 1 de las 3 translocaciones cromosómicas siguientes:
t(8;14).
t(2;8).
t(8;22).
Referencias:
Brearley RL, Johnson SA, Lister TA: Acute lymphoblastic leukaemia in adults: clinicopathological correlations with the French-American-British (FAB) co-operative group classification. Eur J Cancer 15 (6): 909-14, 1979.
Peterson LC, Bloomfield CD, Brunning RD: Blast crisis as an initial or terminal manifestation of chronic myeloid leukemia: a study of 28 patients. Am J Med 60(2): 209-220, 1976.
Secker-Walker LM, Cooke HM, Browett PJ, et al.: Variable Philadelphia breakpoints and potential lineage restriction of bcr rearrangement in acute lymphoblastic leukemia. Blood 72 (2): 784-91, 1988.
Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988.
Sobol RE, Royston I, LeBien TW, et al.: Adult acute lymphoblastic leukemia phenotypes defined by monoclonal antibodies. Blood 65 (3): 730-5, 1985.
Foon KA, Billing RJ, Terasaki PI, et al.: Immunologic classification of acute lymphoblastic leukemia. Implications for normal lymphoid differentiation. Blood 56 (6): 1120-6, 1980.
Información sobre los estadios de la leucemia linfoblástica aguda
No hay ningún sistema de clasificación específico para esta enfermedad; se clasifica como no tratada, en remisión o recidivante.
Leucemia linfoblástica aguda no tratada
En un paciente recién diagnosticado sin tratamiento previo, la LLA no tratada se define de la manera siguiente:
Recuento y diferencial anormal de glóbulos blancos (leucocitos).
Recuentos anómalos de hematocrito o hemoglobina y plaquetas.
Médula ósea anormal con más del 5 % de blastocitos.
Signos y síntomas de la enfermedad.
Leucemia linfoblástica aguda en remisión
Un paciente que ha recibido tratamiento de inducción de la remisión por una LLA está en remisión si se cumplen los criterios que siguen.
La médula ósea es normocelular, con un 5 % de blastocitos o menos.
No hay signos ni síntomas de la enfermedad.
No hay signos ni síntomas de leucemia en el sistema nervioso central ni otra infiltración extramedular.
Todos los valores de laboratorio que siguen están dentro de los límites de referencia:
Recuento y diferencial de glóbulos blancos..
Concentración de hematocrito o hemoglobina.
Recuento plaquetario.
Aspectos generales de las opciones de tratamiento de la leucemia linfoblástica aguda
El tratamiento exitoso de la leucemia linfoblástica aguda (LLA) consiste en controlar la enfermedad sistémica y en la médula ósea, así como el tratamiento (o prevención) de los sitios santuarios, en particular, el sistema nervioso central (SNC).[1,2] La piedra angular de esta estrategia es la quimioterapia combinada administrada de forma sistémica con tratamiento preventivo en el SNC. La profilaxis del SNC se logra con quimioterapia (intratecal o terapia sistémica con dosis alta) y, en algunos casos, radioterapia craneal.
El tratamiento se divide en las siguientes tres fases:
Inducción a la remisión.
Profilaxis del SNC.
Posremisión (también se llama continuación de la remisión o mantenimiento).
La duración promedio del tratamiento para la LLA varía entre 1,5 y 3 años en el esfuerzo de erradicar la población de células leucémicas. Los adultos más jóvenes con LLA tal vez sean idóneos para participar en ciertos ensayos clínicos de LLA infantil. Para obtener más información, consultar la sección Adolescentes y adultos jóvenes con leucemia linfoblástica aguda en Tratamiento de la leucemia linfoblástica aguda infantil.
La participación en un ensayo clínico es muy conveniente, a fin de asegurar un tratamiento adecuado para el paciente y obtener la mayor cantidad de información del tratamiento de esta enfermedad que, si bien responde muy bien a los tratamientos, suele ser mortal.
Cuadro 2. Opciones de tratamiento para la leucemia linfoblástica aguda
Clarkson BD, Gee T, Arlin ZA, et al.: Current status of treatment of acute leukemia in adults: an overview of the Memorial experience and review of literature. Crit Rev Oncol Hematol 4 (3): 221-48, 1986.
Hoelzer D, Gale RP: Acute lymphoblastic leukemia in adults: recent progress, future directions. Semin Hematol 24 (1): 27-39, 1987.
Tratamiento de la leucemia linfoblástica aguda no tratada
Opciones de tratamiento para la leucemia linfoblástica aguda no tratada
Entre las opciones de tratamiento para la leucemia linfoblástica aguda (LLA) no tratada se incluyen las siguientes:
Radioterapia craneal con metotrexato intratecal (IT).
Metotrexato sistémico de dosis elevada y metotrexato IT sin radioterapia craneal.
Quimioterapia IT sola.
Terapia de inducción de la remisión
Es posible que entre el 60 % y el 80 % de los adultos con LLA alcancen un estado de remisión completa después del tratamiento de inducción apropiado, que consiste, por lo general, en un régimen que incluye una combinación de vincristina, prednisona y antraciclinas con asparaginasa o sin esta, lo que produce una tasa de respuesta completa de hasta el 80 %. En pacientes con LLA positiva para Ph, la tasa de remisión es, por lo general, superior al 90 % cuando los regímenes de inducción estándar se combinan con los inhibidores de tirosina–cinasas de BCR::ABL1. En el estudio más grande de pacientes con LLA positiva para Ph publicado hasta la fecha, la supervivencia general (SG) a 5 años de 1913 pacientes adultos con LLA fue del 39 %.[1]
Es habitual que los pacientes que sufren una recaída después de la remisión mueran en el trascurso de 1 año, aun cuando se logre una segunda respuesta completa. Si se dispone de donantes adecuados, y si el paciente es menor de 55 años, tal vez se considere el trasplante de médula ósea para tratar esta enfermedad.[2] Por lo general, los centros de trasplantes que realizan 5 o menos trasplantes al año tienen resultados más precarios que los centros grandes.[3] En lo posible, si se contempla un trasplante alogénico, se recomienda evitar las transfusiones con hemoderivados de un posible donante.[4,5,6,7,8,9,10]
Quimioterapia combinada
La mayoría de los regímenes actuales de inducción para adultos con LLA incluyen prednisona, vincristina y una antraciclina. Algunos regímenes, como aquellos que se usan en el estudio CLB-8811 del Cancer and Leukemia Group (CALGB), también añaden otros fármacos, como la asparaginasa o la ciclofosfamida. Los regímenes de inducción multifarmacológica producen tasas de respuesta completa, que van del 60 % al 90 %.[5,11]; [4,12][[1]]
Mesilato de imatinib
Por lo general, el mesilato de imatinib se incorpora en el plan terapéutico en pacientes con LLA positiva para Ph. El mesilato de imatinib, un inhibidor de tirosina–cinasas de BCR::ABL1 disponible en forma oral, ha mostrado tener actividad clínica en monoterapia para la LLA positiva para Ph.[13,14][Nivel de evidencia C3] Con mayor frecuencia, en particular en pacientes más jóvenes, el imatinib se incorpora en regímenes de quimioterapia combinada. Se han publicado varios estudios de un solo grupo en los que las tasas de respuesta completa y supervivencia se comparan con controles históricos.
Evidencia (mesilato de imatinib):
En varios estudios se indica que añadir imatinib a los regímenes de inducción con quimioterapia convencional combinada da como resultado tasas de respuestas completas, tasas de supervivencia sin complicaciones y tasas de SG más altas que en los controles históricos.[15,16,17] En la actualidad, no es posible llegar a ninguna conclusión a partir de estos estudios, con relación a qué dosis o programa de imatinib resulta óptima.
En un estudio de imatinib combinado con quimioterapia que realizó el Northern Italy Leukemia Group, los pacientes de LLA positiva para Ph recién diagnosticada que no recibieron tratamiento previo se trataron con un régimen de inducción que contenía idarrubicina, vincristina, prednisona y L-asparaginasa.[18] Después de la inscripción de una cohorte inicial, el estudio se modificó para incluir el imatinib (600 mg/d entre los días 15 y 21). En la fase de consolidación, los pacientes recibieron imatinib (600 mg/d durante 7 días) desde 3 días antes del comienzo de cada ciclo de quimioterapia.
La intención fue proceder con un trasplante alogénico para todos aquellos pacientes que lograron la remisión, siempre y cuando se pudiera identificar un donante con compatibilidad de HLA. Los pacientes a los que no se encontraba un donante recibieron un trasplante autógeno. Luego de concluir la quimioterapia y el trasplante, los pacientes debían recibir imatinib como terapia de mantenimiento mientras pudieran tolerarlo. Después de que se logró la participación de 20 pacientes en el grupo de imatinib, se tuvo que omitir la L-asparaginasa del régimen de inducción en ambos grupos debido a su toxicidad.
Los resultados en la primera cohorte de 35 pacientes (sin imatinib) se compararon con aquellos de la cohorte subsiguiente de 59 pacientes (tratados con imatinib). En los pacientes que recibieron imatinib, la probabilidad de SG a 5 años fue del 38 % (mediana de 3,1 años) versus el 23 % en el grupo sin imatinib (mediana de 1,1 años; P = 0,009).[18][Nivel de evidencia C1]
Las deficiencias de este estudio sin aleatorización fueron el tamaño reducido de la muestra (94 pacientes en total) y el cambio en el régimen de tratamiento (omisión de la L-asparaginasa) a mitad del estudio. Sin embargo, los resultados indicaron que la inclusión de imatinib en un régimen de quimioterapia relativamente estándar para pacientes adultos recién diagnosticados con LLA positiva para Ph tal vez proporcione una ventaja significativa de supervivencia.
En otro estudio, 10 pacientes con LLA positiva para Ph y 10 pacientes con leucemia mieloide crónica con crisis blástica linfoide recibieron tratamiento con dosis de imatinib que oscilaban entre 300 y 1000 mg por día.[13] De los 20 pacientes, 4 presentaron remisión hematológica completa y 10 respuestas medulares. Las respuestas fueron de corta duración, y la mayoría de los pacientes experimentaron recaída en la enfermedad al cabo de una mediana de 58 días después del comienzo del tratamiento.
En otro estudio, 48 pacientes con LLA positiva para Ph se trataron con 400 a 800 mg de imatinib por día.[14] La tasa de respuesta general fue del 60 % y 9 de los 48 pacientes (19 %) lograron una remisión completa. Las respuestas, de nuevo, fueron de corta duración, con una mediana de duración de 2,2 meses.
Entre los efectos tóxicos comunes de cada uno de estos estudios se encontraron las náuseas y las anomalías en las enzimas hepáticas, por lo que se necesitó interrumpir o reducir la dosis de imatinib.[13,14] Los trasplantes alogénicos posteriores no se ven afectados de forma adversa por la adición de imatinib al régimen de tratamiento. Para obtener más información, consultar Náuseas y vómitos relacionados con el tratamiento del cáncer.
El imatinib se incorpora, por lo general, al tratamiento de los pacientes con LLA positiva para Ph debido a las respuestas observadas en los ensayos de monoterapia. Si se encuentra un donante adecuado disponible, se deberá considerar el trasplante alogénico de médula ósea porque las remisiones son en general cortas con los ensayos clínicos convencionales de quimioterapia para la LLA.
Cuidados médicos de apoyo
Ya que la mielodepresión es una consecuencia prevista tanto de la leucemia como de su tratamiento quimioterapéutico, los pacientes se deberán vigilar de cerca durante el tratamiento de inducción de la remisión. Deberá haber instalaciones disponibles para apoyo hematológico, así como para el tratamiento de complicaciones infecciosas.
Los cuidados médicos de apoyo durante el tratamiento de inducción de la remisión deberán incluir de forma rutinaria transfusiones de glóbulos rojos y plaquetas cuando resulte apropiado.[19,20]
Evidencia (cuidados médicos de apoyo):
Hay estudios aleatorizados que han mostrado resultados similares en pacientes que recibieron transfusiones profilácticas de plaquetas con concentración de 10 000/mm3 en lugar de 20 000/mm3.[21]
La incidencia de aloinmunización plaquetaría fue similar entre los grupos asignados al azar a recibir uno de los siguientes concentrados de donantes seleccionados al azar:[22]
Concentrados de mezcla de plaquetas.
Concentrados de mezcla de plaquetas filtradas.
Concentrados de mezcla de plaquetas irradiados con rayos ultravioleta B.
Plaquetas filtradas obtenidas por aféresis.
La terapia antimicrobiana empírica de amplio espectro es una necesidad definitiva para los pacientes febriles con neutropenia pronunciada.[23,24] Las instrucciones cuidadosas de higiene personal, cuidado dental y reconocimiento de signos precoces de infección son apropiadas en todos los pacientes. Las instalaciones completas de aislamiento, incluso aire filtrado, comida estéril y esterilización de la flora intestinal no se indican de forma habitual, pero es posible que beneficien a los pacientes que se someten a un trasplante.[25,26]
La ablación rápida de la médula con la consiguiente regeneración temprana de la médula reduce la morbilidad y la mortalidad. Las transfusiones de glóbulos blancos quizás seas beneficiosas para algunos pacientes con médula aplásica e infecciones graves que no responden a los antibióticos.[27] Es posible que los antibióticos orales profilácticos sean apropiados en pacientes en los que se espera una granulocitopenia prolongada y profunda (<100/mm3 durante 2 semanas), aunque son necesarios más estudios.[28] En tales pacientes, es posible que los cultivos de vigilancia en serie sean útiles para detectar la presencia o adquisición de organismos resistentes.
Según se indica en el estudio CALGB (CLB-9111), el uso de factores de crecimiento mieloides durante la terapia de inducción a la remisión disminuye el tiempo para la reconstitución hematopoyética.[29,30]
Terapia profiláctica del sistema nervioso central
La institución temprana de profilaxis del SNC es de suma importancia para lograr el control de la enfermedad que afecta un sitio santuario.
Consideraciones especiales para la leucemia linfoblástica aguda de células B y células T
Hay otros dos subtipos de LLA que requieren una consideración especial. La LLA de células B (que expresa inmunoglobulina de superficie y anomalías citogenéticas, como t(8;14), t(2;8) y t(8;22)), por lo general no se cura con los regímenes típicos para la LLA. Los regímenes radicales muy intensivos de corta duración, como los que se usaron antes en el estudio CLB-9251 (NCT00002494), que son similares a los que se usan en el linfoma no Hodgkin de crecimiento rápido, han mostrado tasas de respuesta y curación elevadas (75 % de respuesta completa; 40 % de supervivencia sin fracaso).[31,32,33] De manera semejante, la LLA de células T, como el linfoma linfoblástico, ha exhibido tasas elevadas de curación cuando se trata con regímenes que contienen ciclofosfamida.[4]
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Goldstone AH, Richards SM, Lazarus HM, et al.: In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood 111 (4): 1827-33, 2008.
Bortin MM, Horowitz MM, Gale RP, et al.: Changing trends in allogeneic bone marrow transplantation for leukemia in the 1980s. JAMA 268 (5): 607-12, 1992.
Horowitz MM, Przepiorka D, Champlin RE, et al.: Should HLA-identical sibling bone marrow transplants for leukemia be restricted to large centers? Blood 79 (10): 2771-4, 1992.
Larson RA, Dodge RK, Burns CP, et al.: A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: cancer and leukemia group B study 8811. Blood 85 (8): 2025-37, 1995.
Linker CA, Levitt LJ, O'Donnell M, et al.: Treatment of adult acute lymphoblastic leukemia with intensive cyclical chemotherapy: a follow-up report. Blood 78 (11): 2814-22, 1991.
Barrett AJ, Horowitz MM, Gale RP, et al.: Marrow transplantation for acute lymphoblastic leukemia: factors affecting relapse and survival. Blood 74 (2): 862-71, 1989.
Dinsmore R, Kirkpatrick D, Flomenberg N, et al.: Allogeneic bone marrow transplantation for patients with acute lymphoblastic leukemia. Blood 62 (2): 381-8, 1983.
Jacobs AD, Gale RP: Recent advances in the biology and treatment of acute lymphoblastic leukemia in adults. N Engl J Med 311 (19): 1219-31, 1984.
Doney K, Buckner CD, Kopecky KJ, et al.: Marrow transplantation for patients with acute lymphoblastic leukemia in first marrow remission. Bone Marrow Transplant 2 (4): 355-63, 1987.
Vernant JP, Marit G, Maraninchi D, et al.: Allogeneic bone marrow transplantation in adults with acute lymphoblastic leukemia in first complete remission. J Clin Oncol 6 (2): 227-31, 1988.
Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988.
Kantarjian H, Thomas D, O'Brien S, et al.: Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 101 (12): 2788-801, 2004.
Druker BJ, Sawyers CL, Kantarjian H, et al.: Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 344 (14): 1038-42, 2001.
Ottmann OG, Druker BJ, Sawyers CL, et al.: A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias. Blood 100 (6): 1965-71, 2002.
Thomas DA, Faderl S, Cortes J, et al.: Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 103 (12): 4396-407, 2004.
Yanada M, Takeuchi J, Sugiura I, et al.: High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 24 (3): 460-6, 2006.
Wassmann B, Pfeifer H, Goekbuget N, et al.: Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood 108 (5): 1469-77, 2006.
Bassan R, Rossi G, Pogliani EM, et al.: Chemotherapy-phased imatinib pulses improve long-term outcome of adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: Northern Italy Leukemia Group protocol 09/00. J Clin Oncol 28 (22): 3644-52, 2010.
Slichter SJ: Controversies in platelet transfusion therapy. Annu Rev Med 31: 509-40, 1980.
Murphy MF, Metcalfe P, Thomas H, et al.: Use of leucocyte-poor blood components and HLA-matched-platelet donors to prevent HLA alloimmunization. Br J Haematol 62 (3): 529-34, 1986.
Rebulla P, Finazzi G, Marangoni F, et al.: The threshold for prophylactic platelet transfusions in adults with acute myeloid leukemia. Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto. N Engl J Med 337 (26): 1870-5, 1997.
Leukocyte reduction and ultraviolet B irradiation of platelets to prevent alloimmunization and refractoriness to platelet transfusions. The Trial to Reduce Alloimmunization to Platelets Study Group. N Engl J Med 337 (26): 1861-9, 1997.
Hughes WT, Armstrong D, Bodey GP, et al.: From the Infectious Diseases Society of America. Guidelines for the use of antimicrobial agents in neutropenic patients with unexplained fever. J Infect Dis 161 (3): 381-96, 1990.
Rubin M, Hathorn JW, Pizzo PA: Controversies in the management of febrile neutropenic cancer patients. Cancer Invest 6 (2): 167-84, 1988.
Armstrong D: Symposium on infectious complications of neoplastic disease (Part II). Protected environments are discomforting and expensive and do not offer meaningful protection. Am J Med 76 (4): 685-9, 1984.
Sherertz RJ, Belani A, Kramer BS, et al.: Impact of air filtration on nosocomial Aspergillus infections. Unique risk of bone marrow transplant recipients. Am J Med 83 (4): 709-18, 1987.
Schiffer CA: Granulocyte transfusions: an overlooked therapeutic modality. Transfus Med Rev 4 (1): 2-7, 1990.
Wade JC, Schimpff SC, Hargadon MT, et al.: A comparison of trimethoprim-sulfamethoxazole plus nystatin with gentamicin plus nystatin in the prevention of infections in acute leukemia. N Engl J Med 304 (18): 1057-62, 1981.
Scherrer R, Geissler K, Kyrle PA, et al.: Granulocyte colony-stimulating factor (G-CSF) as an adjunct to induction chemotherapy of adult acute lymphoblastic leukemia (ALL). Ann Hematol 66 (6): 283-9, 1993.
Larson RA, Dodge RK, Linker CA, et al.: A randomized controlled trial of filgrastim during remission induction and consolidation chemotherapy for adults with acute lymphoblastic leukemia: CALGB study 9111. Blood 92 (5): 1556-64, 1998.
Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
Lee EJ, Petroni GR, Schiffer CA, et al.: Brief-duration high-intensity chemotherapy for patients with small noncleaved-cell lymphoma or FAB L3 acute lymphocytic leukemia: results of cancer and leukemia group B study 9251. J Clin Oncol 19 (20): 4014-22, 2001.
Thomas DA, Cortes J, O'Brien S, et al.: Hyper-CVAD program in Burkitt's-type adult acute lymphoblastic leukemia. J Clin Oncol 17 (8): 2461-70, 1999.
Tratamiento de la leucemia linfoblástica aguda en remisión
Opciones de tratamiento de la leucemia linfoblástica aguda en remisión
Entre las opciones de tratamiento para la leucemia linfoblástica aguda (LLA) en remisión se incluyen las siguientes:
Radioterapia craneal con metotrexato intratecal (IT).
Metotrexato sistémico de dosis elevada y metotrexato IT sin radioterapia craneal.
Quimioterapia IT sola.
Terapia de posremisión
Los abordajes actuales para la terapia de posremisión para la LLA incluyen quimioterapia breve relativamente intensiva, seguida de cualquiera de los siguientes procedimientos:
Tratamiento a largo plazo con dosis más bajas (terapia de mantenimiento).
En varios ensayos, incluidos los del Cancer and Leukemia Group B (CLB-8811) y el European Cooperative Oncology Group (ECOG-2993 [NCT00002514]), de quimioterapia posremisión intensiva para LLA, se ha confirmado una tasa de supervivencia sin enfermedad (SSE) a largo plazo de alrededor del 40 %.[1,2,3,4,5,6,7]
En dos series,[4,5] se encontraron particularmente buenos pronósticos para pacientes de LLA con linaje de células T, con tasas de SSE del 50 % al 70 % para pacientes que recibieron terapia de posremisión.
Estas series representan una mejoría significativa en las tasas de SSE, en comparación con abordajes quimioterapéuticos anteriores menos intensivos.
Por el contrario, se mostraron tasas de curación precarias en pacientes con LLA positiva para el cromosoma Filadelfia (Ph), LLA con linaje de células B con un fenotipo L3 (positivo para inmunoglobulina de superficie) y LLA con linaje de células B caracterizada por t(4;11).
La administración de los programas de dosis intensivas más nuevos puede ser difícil y se debe efectuar por médicos expertos en estos regímenes en centros equipados para tratar posibles complicaciones. Los estudios en los que se eliminó la quimioterapia de continuación o de mantenimiento tuvieron resultados inferiores a aquellos con duraciones prolongadas de tratamiento.[8,9] El imatinib se ha incorporado en los regímenes de mantenimiento de pacientes con LLA positiva para Ph.[10,11,12]
Evidencia (TMO alogénico y autógeno):
El TMO alogénico produce una incidencia más baja de recaída leucémica, aunque se compare con el trasplante de médula ósea de un gemelo monocigótico (TMO singénico). Este resultado ha conducido al concepto de un efecto inmunológico de injerto contra leucemia, similar a la enfermedad de injerto contra huésped (EICH). La mejoría en cuanto a la SSE en pacientes que se someten a TMO alogénico como terapia primaria posremisión se ve afectada, en parte, por el aumento de la morbilidad y la mortalidad de EICH, enfermedad venooclusiva del hígado y neumonitis intersticial.[13]
Los resultados de una serie de estudios retrospectivos y prospectivos publicados entre 1987 y 1994 indican que el TMO alogénico o el TMO autógeno, como terapias de posremisión, no ofrecen ventaja en cuanto a la supervivencia sobre la quimioterapia intensiva salvo, quizás, para pacientes con LLA de riesgo alto positiva para Ph.[14,15,16,17] Esto se confirmó en el estudio ECOG-2993 (NCT01505699).[7]
El uso de TMO alogénico como terapia primaria de posremisión está limitado tanto por la necesidad de un donante fraterno con compatibilidad de HLA como por el aumento de la mortalidad a causa del TMO alogénico en pacientes en su quinta o sexta década de vida.
La mortalidad por TMO alogénico con un donante fraterno con compatibilidad en estos estudios osciló entre el 20 % y el 40 %.
A partir de los resultados de los estudios anteriores, se puso en marcha un ensayo internacional de LLA (ECOG-2993) con la intención de examinar de manera más definitiva la función del trasplante como terapia de posremisión para la LLA; se inscribieron pacientes entre 1993 a 2006.[7] Los pacientes con LLA negativa para el Ph entre los 15 a 59 años de edad recibieron tratamiento de inducción multifarmacológico idéntico que se asemejaba a regímenes publicados con anterioridad.[1,2,3] A continuación, los pacientes en remisión se catalogaron como idóneos para la tipificación de HLA; los pacientes con un donante fraterno con compatibilidad completa se sometieron a TMO alogénico como terapia de consolidación. Los pacientes sin donantes se asignaron al azar para recibir TMO autógeno o quimioterapia de mantenimiento. El desenlace primario medido fue la supervivencia general (SG) y los desenlaces secundarios fueron la supervivencia sin complicaciones, la tasa de recidiva y la mortalidad sin recaída. Se inscribieron 1929 pacientes, que se estratificaron de acuerdo con la edad, el recuento de leucocitos y el tiempo que tardó en aparecer la recidiva. Los pacientes con riesgo alto se definieron como aquellos con un recuento de glóbulos blancos alto en el momento de la presentación, o aquellos mayores de 35 años.
De los participantes en este estudio, el 90 % logró la remisión tras el tratamiento de inducción y, de estos pacientes, se encontró que 443 tenían un hermano con HLA idénticos; 310 de ellos se sometieron a TMO alogénico. De los 456 pacientes en remisión idóneos para el trasplante, pero que carecían de donante, 227 recibieron quimioterapia sola, mientras que 229 se sometieron a un TMO autógeno.
En un análisis pacientes con donantes y sin donantes, los pacientes con LLA de riesgo estándar que tenían un hermano con HLA idénticos, tuvieron una tasa de SG a 5 años del 53 %, en comparación con el 45 % en los pacientes que carecían de donante (P = 0,01).
En un análisis de subgrupos, la ventaja de los pacientes con donantes siguió siendo significativa en pacientes con LLA de riesgo estándar (tasa de SG = 62 vs. 52 %; P = 0,02).
En los pacientes con enfermedad de riesgo alto (mayores de 35 años o con un recuento de glóbulos blancos alto), la diferencia en cuanto a la SG fue del 41 vs. el 35 % (donante vs. no donante), pero no fue significativa (P = 0,2).
Las tasas de recaída fueron significativamente inferiores (P < 0,00005) tanto para los pacientes estándar como para los de riesgo alto con donante con compatibilidad de HLA.
En contraste con el TMO alogénico, el TMO autógeno fue menos eficaz que el tratamiento de mantenimiento como terapia de posremisión (la tasa de SG a 5 años fue del 46 % para la quimioterapia vs. el 37 % para el TMO autógeno; P = 0,03).
Los resultados de este ensayo indican un efecto injerto contra leucemia en los adultos con LLA negativa para el Ph, y respalda el uso del TMO alogénico de donante fraterno como tratamiento de consolidación que proporciona la mayor probabilidad de supervivencia a largo plazo para la LLA en adultos de riesgo estándar en su primera remisión.[7][Nivel de evidencia B4]
Los resultados también indicaron que, ante la ausencia de donante fraterno, es preferible la quimioterapia de mantenimiento al TMO autógeno como terapia de remisión.[7][Nivel de evidencia B4]
El uso de donantes compatibles no emparentados para un TMO alogénico se encuentra en evaluación, pero debido a la alta morbilidad y mortalidad relacionas con el tratamiento, se reserva para los pacientes en segunda o posteriores remisiones. La dosis administrada de irradiación corporal total se relaciona con la incidencia de EICH crónica y aguda, y es posible que sea un predictor independiente de la supervivencia sin leucemia.[18][Nivel de evidencia C1]
Evidencia (LLA de células B):
Los regímenes intensivos a base de ciclofosfamida similares a los que se usan en el linfoma no Hodgkin de crecimiento rápido han mostrado una mejoría en los resultados de la SSE para pacientes con LLA de células B (tipo morfológico L3, positiva para la inmunoglobulina de superficie).[19]
Un grupo de investigadores hizo una revisión retrospectiva de tres ensayos secuenciales de grupos cooperativos de Alemaniay se encontró lo siguiente:[19]
Una mejoría marcada en la supervivencia, de 0 sobrevivientes en un estudio de 1981 en el que se usó tratamiento pediátrico estándar y que duró 2,5 años, a una tasa de supervivencia del 50 % en 2 ensayos posteriores en los que se usó quimioterapia alternada rápidamente como para el linfoma y completada en 6 meses.
Terapia profiláctica del sistema nervioso central
La institución temprana de profilaxis del SNC es de suma importancia para lograr el control de la enfermedad que afecta un sitio santuario. Algunos autores han indicado que hay un subgrupo de pacientes con riesgo bajo de recaída en el SNC para quienes es posible que la profilaxis del SNC no sea necesaria. No obstante, este concepto no se ha probado de manera prospectiva.[20]
La profilaxis intensiva del SNC sigue siendo un componente prominente del tratamiento.[19] Este informe, que requiere confirmación en otros entornos de grupos cooperativos, es alentador para los pacientes con LLA L3. Los pacientes con inmunoglobulina de superficie y tipo morfológico L1 o L2 no se beneficiaron de este régimen. En forma semejante, los pacientes con tipo morfológico e inmunofenotipo L3, pero con características citogenéticas atípicas, no se curaron con este abordaje. En un análisis univariante, un recuento de glóbulos blancos de menos de 50 000 por microlitro predijo una mejora de la supervivencia sin leucemia.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Gaynor J, Chapman D, Little C, et al.: A cause-specific hazard rate analysis of prognostic factors among 199 adults with acute lymphoblastic leukemia: the Memorial Hospital experience since 1969. J Clin Oncol 6 (6): 1014-30, 1988.
Hoelzer D, Thiel E, Löffler H, et al.: Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood 71 (1): 123-31, 1988.
Linker CA, Levitt LJ, O'Donnell M, et al.: Treatment of adult acute lymphoblastic leukemia with intensive cyclical chemotherapy: a follow-up report. Blood 78 (11): 2814-22, 1991.
Zhang MJ, Hoelzer D, Horowitz MM, et al.: Long-term follow-up of adults with acute lymphoblastic leukemia in first remission treated with chemotherapy or bone marrow transplantation. The Acute Lymphoblastic Leukemia Working Committee. Ann Intern Med 123 (6): 428-31, 1995.
Larson RA, Dodge RK, Burns CP, et al.: A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: cancer and leukemia group B study 8811. Blood 85 (8): 2025-37, 1995.
Kantarjian H, Thomas D, O'Brien S, et al.: Long-term follow-up results of hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone (Hyper-CVAD), a dose-intensive regimen, in adult acute lymphocytic leukemia. Cancer 101 (12): 2788-801, 2004.
Goldstone AH, Richards SM, Lazarus HM, et al.: In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood 111 (4): 1827-33, 2008.
Cuttner J, Mick R, Budman DR, et al.: Phase III trial of brief intensive treatment of adult acute lymphocytic leukemia comparing daunorubicin and mitoxantrone: a CALGB Study. Leukemia 5 (5): 425-31, 1991.
Dekker AW, van't Veer MB, Sizoo W, et al.: Intensive postremission chemotherapy without maintenance therapy in adults with acute lymphoblastic leukemia. Dutch Hemato-Oncology Research Group. J Clin Oncol 15 (2): 476-82, 1997.
Thomas DA, Faderl S, Cortes J, et al.: Treatment of Philadelphia chromosome-positive acute lymphocytic leukemia with hyper-CVAD and imatinib mesylate. Blood 103 (12): 4396-407, 2004.
Yanada M, Takeuchi J, Sugiura I, et al.: High complete remission rate and promising outcome by combination of imatinib and chemotherapy for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia: a phase II study by the Japan Adult Leukemia Study Group. J Clin Oncol 24 (3): 460-6, 2006.
Wassmann B, Pfeifer H, Goekbuget N, et al.: Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood 108 (5): 1469-77, 2006.
Horowitz MM, Messerer D, Hoelzer D, et al.: Chemotherapy compared with bone marrow transplantation for adults with acute lymphoblastic leukemia in first remission. Ann Intern Med 115 (1): 13-8, 1991.
Sebban C, Lepage E, Vernant JP, et al.: Allogeneic bone marrow transplantation in adult acute lymphoblastic leukemia in first complete remission: a comparative study. French Group of Therapy of Adult Acute Lymphoblastic Leukemia. J Clin Oncol 12 (12): 2580-7, 1994.
Forman SJ, O'Donnell MR, Nademanee AP, et al.: Bone marrow transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood 70 (2): 587-8, 1987.
Fière D, Lepage E, Sebban C, et al.: Adult acute lymphoblastic leukemia: a multicentric randomized trial testing bone marrow transplantation as postremission therapy. The French Group on Therapy for Adult Acute Lymphoblastic Leukemia. J Clin Oncol 11 (10): 1990-2001, 1993.
Corvò R, Paoli G, Barra S, et al.: Total body irradiation correlates with chronic graft versus host disease and affects prognosis of patients with acute lymphoblastic leukemia receiving an HLA identical allogeneic bone marrow transplant. Int J Radiat Oncol Biol Phys 43 (3): 497-503, 1999.
Hoelzer D, Ludwig WD, Thiel E, et al.: Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood 87 (2): 495-508, 1996.
Kantarjian HM, Walters RS, Smith TL, et al.: Identification of risk groups for development of central nervous system leukemia in adults with acute lymphocytic leukemia. Blood 72 (5): 1784-9, 1988.
Tratamiento de la leucemia linfoblástica aguda recidivante
Opciones de tratamiento para la leucemia linfoblástica aguda recidivante
Entre las opciones de tratamiento para la leucemia linfoblástica aguda (LLA) recidivante se incluyen las siguientes:
Dasatinib (para pacientes con LLA positivo para el cromosoma Filadelfia [Ph]).
Los pacientes que no tienen donante con compatibilidad de HLA son excelentes candidatos para participar en ensayos clínicos en los que se estudian los siguientes tratamientos:[1,2,3,4,5,6,7]
Trasplante autógeno.
Inmunomodulación.
Terapia de células T con receptor de antígeno quimérico (CAR).[8]
Fármacos quimioterapéuticos o biológicos novedosos.
Quimioterapia de reinducción seguida de trasplante de médula ósea alogénico
Los pacientes con LLA que presentan recidiva después de la quimioterapia y el tratamiento de mantenimiento son difíciles de curar mediante quimioterapia adicional sola. Estos pacientes se deben considerar para la quimioterapia de reinducción seguida de TMO alogénico.
Blinatumomab seguido de trasplante de médula ósea alogénico
El blinatumomab es un anticuerpo biespecífico dirigido a CD19 y CD3. La Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) aprobó el uso de blinatumomab en pacientes con LLA de células B en recaída o resistente al tratamiento.
Evidencia (blinatumomab):
Se realizó un estudio aleatorizado de fase III sobre blinatumomab versus 1 de 4 regímenes de reinducción estándar en pacientes con enfermedad primaria resistente al tratamiento, que fue resistente a la terapia de rescate, con una primera recaída que duró menos de 12 meses, una segunda o posteriores recaídas, o cualquier recaída tras un trasplante alogénico.[9] Los 4 regímenes incluyeron lo siguiente: fludarabina; arabinósido de citosina en dosis altas y factor estimulante de colonias de granulocitos, con antraciclina o sin esta; un régimen a base de dosis altas de arabinósido de citosina; un régimen a base de dosis altas de metotrexato; o un régimen a base de clofarabina.
Las tasas de remisión para el grupo tratado con blinatumomab fueron del 43,9 % vs. el 24,6 % para el grupo de tratamiento estándar (oportunidad relativa 2,40, intervalo de confianza [IC] 95 %, 1,51–3,80).
La supervivencia general (SG) fue superior para el grupo tratado con blinatumomab (7,7 meses vs. 4,0 meses en el grupo de tratamiento estándar) con un cociente de riesgos instantáneos (CRI) de 0,71 (IC 95 %, 0,55–0,93) en favor del blinatumomab.
Los efectos adversos fueron similares en ambos grupos; el único efecto secundario distintivo del blinatumomab fue el síndrome de liberación de citocinas observado en el 4,9 % de los pacientes.
El blinatumomab se debe considerar como terapia estándar de reinducción para aquellos pacientes con enfermedad primaria resistente al tratamiento, que es resistente a la terapia de rescate, con una primera recaída que duró menos de 12 meses, una segunda o posteriores recaídas, o cualquier recaída tras un trasplante alogénico.[9][Nivel de evidencia A1]
Inotuzumab ozogamicina seguido de trasplante de médula ósea alogénico
El inotuzumab ozogamicina es un conjugado anticuerpo-fármaco dirigido a CD22 que contiene caliqueamicina, una toxina conjugada. La FDA ha aprobado el inotuzumab ozogamicina para su administración a pacientes con LLA de células B que expresan CD22, en recaída o resistente al tratamiento.
Evidencia (inotuzumab ozogamicina):
En un estudio aleatorizado de fase III, en el que se comparó el inotuzumab ozogamicina versus 1 de los 3 regímenes de reinducción estándar, se incluyó a 218 pacientes de 18 años o más con enfermedad recidivante o resistente al tratamiento, que se iban a someter a su primer o segundo régimen de rescate.[10] Los 3 regímenes estándar contenían fludarabina, citarabina y factor estimulante de colonias de granulocitos (FLAG), citarabina y mitoxantrona, o dosis altas de citarabina.
Las tasas de remisión completa o de remisión completa con recuperación de recuento incompleto fueron del 80,7 % (IC 95 %, 72,1–87,7 %) en el grupo de inotuzumab versus el 29,4 % (IC 95 %, 21,0–38,8 %) en el grupo de tratamiento estándar (P < 0,001).
La supervivencia sin progresión fue superior en el grupo que recibió tratamiento con inotuzumab (5,0 vs. 1,8 meses en el grupo de tratamiento estándar) con un CRI de 0,45 (IC 97,5 %, 0,34–0,61; P < 0,001).
La duración de la remisión fue breve en ambos grupos: en el grupo de inotuzumab fue de 4,6 meses (IC 95 %, 3,9–5,4) y en el grupo de tratamiento estándar fue de 3,1 meses (IC 95 %, 1,4–4,9).
De los 48 pacientes del grupo de inotuzumab que se sometieron a un trasplante después del tratamiento, 10 presentaron enfermedad venooclusiva. De los 20 pacientes del grupo de quimioterapia estándar que se sometieron a un trasplante después de la remisión, 1 paciente presentó enfermedad venooclusiva.
La SG no fue prolongada desde el punto de vista estadístico en el grupo de inotuzumab (7,7 meses en el grupo de inotuzumab vs. 6,7 meses en el grupo de tratamiento estándar (CRI, 0,77; IC 97,5 %, 0,58–1,03; P = 0,04) debido al fracaso de alcanzar un límite de P = 0,0208 prespecificado en el estudio.
Los efectos adversos (≥grado 3) que afectaron el hígado fueron más altos en el grupo de inotuzumab: el 11 % de los pacientes presentaron enfermedad venooclusiva hepática, en comparación con el 1 % de los pacientes del grupo de tratamiento estándar.
El inotuzumab ozogamicina se debe considerar como una opción para la reinducción en pacientes con LLA con expresión de CD22 recidivante o resistente al tratamiento.[10][Nivel de evidencia B1]
Radioterapia paliativa
Se podrían considerar dosis bajas de radioterapia paliativa entre los pacientes con recidiva sintomática, ya sea dentro o fuera del sistema nervioso central.[11]
Dasatinib
Con frecuencia, los pacientes con LLA positiva para Ph reciben imatinib en el momento de la recaída y, por tanto, tendrán enfermedad resistente a este fármaco. El dasatinib es un nuevo inhibidor de tirosina–cinasas con eficacia contra varias mutaciones de genes de fusión BCR::ABL1. Se ha aprobado su uso en pacientes con LLA positiva para Ph que son resistentes o intolerantes al imatinib. Dicha aprobación se basó en una serie de ensayos con pacientes con leucemia mieloide crónica, uno de los cuales incluyó un número pequeño de pacientes con crisis blástica linfoide o LLA positiva para Ph.
Evidencia (dasatinib):
En un estudio, 10 pacientes se trataron con dasatinib en dosis escalonadas.[12] Se observó en 7 de estos pacientes una respuesta hematológica completa (<5 % de blastocitos medulares con recuentos de sangre periférica normales); 3 de ellos tuvieron una respuesta citogenética completa.
Los efectos tóxicos comunes consistieron en mielodepresión reversible (89 %) y derrames pleurales (21 %).
Casi todos estos pacientes tuvieron recaídas en el trascurso de 6 meses a partir del tratamiento con dasatinib.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Herzig RH, Bortin MM, Barrett AJ, et al.: Bone-marrow transplantation in high-risk acute lymphoblastic leukaemia in first and second remission. Lancet 1 (8536): 786-9, 1987.
Thomas ED, Sanders JE, Flournoy N, et al.: Marrow transplantation for patients with acute lymphoblastic leukemia: a long-term follow-up. Blood 62 (5): 1139-41, 1983.
Barrett AJ, Horowitz MM, Gale RP, et al.: Marrow transplantation for acute lymphoblastic leukemia: factors affecting relapse and survival. Blood 74 (2): 862-71, 1989.
Dinsmore R, Kirkpatrick D, Flomenberg N, et al.: Allogeneic bone marrow transplantation for patients with acute lymphoblastic leukemia. Blood 62 (2): 381-8, 1983.
Sallan SE, Niemeyer CM, Billett AL, et al.: Autologous bone marrow transplantation for acute lymphoblastic leukemia. J Clin Oncol 7 (11): 1594-601, 1989.
Paciucci PA, Keaveney C, Cuttner J, et al.: Mitoxantrone, vincristine, and prednisone in adults with relapsed or primarily refractory acute lymphocytic leukemia and terminal deoxynucleotidyl transferase positive blastic phase chronic myelocytic leukemia. Cancer Res 47 (19): 5234-7, 1987.
Biggs JC, Horowitz MM, Gale RP, et al.: Bone marrow transplants may cure patients with acute leukemia never achieving remission with chemotherapy. Blood 80 (4): 1090-3, 1992.
Maude SL, Frey N, Shaw PA, et al.: Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371 (16): 1507-17, 2014.
Kantarjian H, Stein A, Gökbuget N, et al.: Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med 376 (9): 836-847, 2017.
Kantarjian HM, DeAngelo DJ, Stelljes M, et al.: Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 375 (8): 740-53, 2016.
Gray JR, Wallner KE: Reversal of cranial nerve dysfunction with radiation therapy in adults with lymphoma and leukemia. Int J Radiat Oncol Biol Phys 19 (2): 439-44, 1990.
Talpaz M, Shah NP, Kantarjian H, et al.: Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 354 (24): 2531-41, 2006.
Actualizaciones más recientes a este resumen (03 / 26 / 2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se actualizaron las estadísticas con el número estimado de casos nuevos y defunciones para 5 (se citó a la American Cancer Society como referencia 1).
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento de la leucemia linfoblástica aguda en adultos. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento para adultos, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
El revisor principal del sumario sobre Tratamiento de la leucemia linfoblástica aguda es:
Aaron Gerds, MD (Cleveland Clinic Taussig Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento para adultos emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.