Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Introducción
La leiomiomatosis hereditaria y cáncer de células renales (HLRCC) se caracteriza por la presencia de una o más de las siguientes lesiones: leiomiomas cutáneos (leiomiomatosis), leiomiomas uterinos (fibromas) en mujeres, y cáncer de células renales (RCC). Las variantes patogénicas germinales del gen FH son la causa de la susceptibilidad a la HLRCC. El gen FH codifica la fumarato–hidratasa, una enzima que cataliza la conversión de fumarato a malato en el ciclo del ácido tricarboxílico (ciclo de Krebs).
Nomenclatura
Tradicionalmente, la predisposición al desarrollo de leiomiomas cutáneos se llamó leiomiomatosis cutánea múltiple. En 1973, se describieron 2 familias extensas con varios miembros en más de 3 generaciones afectados por leiomiomas cutáneos y leiomiomas o leiomiosarcomas uterinos de herencia autosómica dominante.[1] También se notificó el caso de una mujer de 20 años con leiomiosarcoma uterino y RCC metastásico. Posteriormente, la asociación de leiomiomas cutáneos y leiomiomas uterinos se llamó síndrome de Reed. Sin embargo, no se describió un vínculo claro entre los leiomiomas cutáneos y el RCC hasta 2001, cuando en un estudio se notificaron 2 familias finlandesas que exhibieron cosegregación de leiomiomas cutáneos, leiomiomas uterinos y RCC papilar de tipo 2;[2] a partir de ese momento se introdujeron nombres como leiomiomatosis hereditaria y cáncer de células renales (HLRCC) o el síndrome de leiomiomatosis hereditaria asociado a carcinoma de células renales.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Características genéticas
GenFH
El gen FH consta de 10 exones que contienen 22,15 kb de DNA. Es un gen muy conservado en todas las especies. El gen FH humano se encuentra en el cromosoma 1q42.3-43.
La leiomiomatosis hereditaria y cáncer de células renales (HLRCC) es un síndrome de herencia autosómica dominante. Una persona presenta una predisposición a manifestar la HLRCC cuando hereda una variante patogénica heterocigota en el gen FH.[1] Se han identificado diferentes variantes patogénicas en familias con HLRCC. En su mayoría son variantes de cambio de sentido, pero también se han identificado variantes sin sentido, cambios en el marco de lectura, variantes del sitio de empalme y deleciones parciales o completas del gen.[2,3,4,5,6]
Los tumores renales que se presentan en personas que heredan una variante patogénica germinal de FH por lo general exhiben pérdida de heterocigosidad debido a una segunda variante somática de FH. Este hallazgo indica que la pérdida de la función de la proteína fumarato–hidratasa es la causa de la formación de tumores en la HLRCC, y respalda la teoría de que FH cumple una función como supresor de tumores.[2,7]
Cuando se heredan variantes patogénicas bialélicas en el gen FH (homocigosis o heterocigosis compuesta) es posible que se produzca una deficiencia de fumarato–hidratasa (FHD) de herencia autosómica recesiva (FMRD), un trastorno caracterizado por alteración neurológica neonatal que progresa con rapidez, y que incluye hipotonía, convulsiones y atrofia cerebral. Para obtener más información, consultar la sección Trastornos relacionados genéticamente.
Prevalencia
Los datos estimados sobre la prevalencia de HLRCC continúan en evolución. Los datos más antiguos indican una prevalencia de 1 en cada 200 000 personas.[8] Sin embargo, en un análisis de cohortes no poblacionales se encontró una frecuencia de portadores de FH de 1 en cada 1000 personas, lo que indica un mayor número de casos asintomáticos de lo que se pensaba.[9] En un análisis de una base de datos clínica de más de 120 000 registros de personas que se sometieron a pruebas genéticas de cáncer hereditario se estimó que la frecuencia de portadores era de 1 cada 2668 (0,04 %). Estas estimaciones reflejan la tasa alta de variantes patogénicas de FH detectada de manera indirecta en personas que se sometieron a pruebas genéticas por otras indicaciones. Estos hallazgos indican que quizás la HLRCC sea uno de los síndromes de cáncer hereditario más frecuentes.[10]
Penetrancia de variantes patogénicas deFH
A partir de la observación de que la mayoría de los pacientes con HLRCC presentan al menos 1 de las 3 manifestaciones clínicas principales, se considera que la penetrancia de HLRCC en portadores de variantes patogénicas de FH es muy alta. Sin embargo, el cálculo de la incidencia acumulada durante la vida del cáncer de células renales (RCC) varía mucho, y la mayoría de las estimaciones oscilan del 15 al 30 % en familias con variantes patogénicas germinales de FH, según el método de comprobación y las imágenes utilizadas.[3,4,7,11,12,13] En un estudio de 2020 se observó que la tasa de variantes patogénicas de FH detectadas era más elevada de lo que se pensaba, lo que indica que el riesgo de por vida de cáncer de riñón en personas con HLRCC quizás sea significativamente inferior a las estimaciones vigentes.[9]
En un estudio en el que se analizaron pacientes que se sometieron a pruebas genéticas se indicó que el RCC quizás sea más frecuente en hombres con HLRCC que en mujeres con HLRCC.[10] En varios estudios también se notificó un aumento de variantes germinales patogénicas de FH en personas con RCC y ascendencia africana.[10,14]
Correlaciones entre genotipo y fenotipo
No se han notificado correlaciones entre variantes de FH específicas y la aparición de lesiones cutáneas, leiomiomas uterinos, RCC u otras características principales de la HLRCC.[4]
Sin embargo, ciertas variantes de FH (sobre todo variantes de cambio de sentido) se relacionan con un aumento del riesgo de paraganglioma o feocromocitoma en ausencia de otras características de HLRCC.[15] En la literatura se han notificado 3 variantes recurrentes que aumentan el riesgo de paraganglioma o feocromocitoma: 1) p.Thr234Ala, 2) p.Ala273Thr y 3) p.Ala194Thr. Por el contrario, las variantes de FH relacionadas con HLRCC autosómica dominante y FHD autosómica recesiva no se correlacionan con un aumento del riesgo de paraganglioma o feocromocitoma. A pesar de que la vigilancia de paraganglioma o feocromocitomas no se incluye en las pautas para la HLRCC, los autores indican que se debe garantizar una vigilancia similar a la que se recomienda para los portadores de SDHx. Para obtener más información, consultar la sección Vigilancia.
Análisis de secuencia
Con el uso de métodos de secuenciación bidireccional de DNA, se han detectado variantes patogénicas de FH en más del 85 % de las personas con HLRCC.[3,4,16]
Trastornos relacionados genéticamente
Deficiencia de fumarato–hidratasa
La deficiencia de fumarato–hidratasa (aciduria fumárica, FHD, FMRD) es un trastorno autosómico recesivo que se produce por la herencia de variantes patogénicas bialélicas del gen FH. Este problema metabólico congénito se caracteriza por un deterioro neurológico de progresión rápida que incluye hipotonía, convulsiones y atrofia cerebral. En personas con FHD se encuentran variantes patogénicas germinales de FH homocigóticas y heterocigóticas compuestas.[17,18] Hasta la fecha, no se ha notificado RCC en personas afectadas por FHD, quizás porque la mayoría de las personas con este trastorno mueren a los pocos meses de vida y muy pocos llegan a la edad adulta temprana.[19] Sin embargo, se notificó que un progenitor (portador heterocigoto) de una persona con FHD presentó leiomiomas cutáneos similares a los observados en la HLRCC.[2]
Se cree que hay 4 variantes de FH relacionadas con la FHD: 1) p.Lys477dup, 2) p.Pro174Arg, 3) p.Gln376Pro y 4) p.Ala308Gly.[20] La variante que se encuentra con más frecuencia en las personas con FHD es la duplicación dentro del marco de lectura, c.1431_1433dupAAA (también conocida como p.Lys477dup). Esta variante se ha observado en el 0,5% de la población judía asquenazí.
Si bien se ha demostrado en múltiples estudios que ciertas variantes patogénicas bialélicas pueden causar FHD autosómica recesiva, estas variantes no están relacionadas con la HLRCC cuando se presentan en estado heterocigótico.[10,20] Por lo tanto, no se cree que la variante de FHD común, p.Lys477dup, se relacione con un aumento del riesgo de RCC. Sin embargo, la variante p.Lys477dup está asociada a la FHD autosómica recesiva cuando se encuentra con otra variante de FH (es decir, p.Lys477dup en un alelo y una variante de FH diferente en otro alelo), y no cuando se presenta en estado homocigótico (es decir, cuando la persona es portadora de 2 variantes p.Lys477dup). Este hallazgo indica que la variante p.Lys477dup tiene cierta funcionalidad. En estos estudios, las personas portadoras de la variante de FH p.Lys477dup, que además tenían RCC, presentabas características similares a las de las personas que tenían formas esporádicas de cáncer de riñón, como una mayor prevalencia de RCC de células claras y una presentación del RCC a edades más avanzadas.
Variantes somáticas deFH
Se identificó la pérdida somática bialélica de FH en 2 casos de leiomiomas uterinos esporádicos de inicio temprano y 1 caso de sarcoma de tejido blando del miembro inferior en personas sin otras características tumorales asociadas a la enfermedad hereditaria.[21,22] En las formas esporádicas de RCC se han identificado pocas variantes somáticas de FH.[21,23]
Referencias:
Alam NA, Rowan AJ, Wortham NC, et al.: Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12 (11): 1241-52, 2003.
Tomlinson IP, Alam NA, Rowan AJ, et al.: Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30 (4): 406-10, 2002.
Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Wei MH, Toure O, Glenn GM, et al.: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43 (1): 18-27, 2006.
Bayley JP, Launonen V, Tomlinson IP: The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet 9: 20, 2008.
Vocke CD, Ricketts CJ, Merino MJ, et al.: Comprehensive genomic and phenotypic characterization of germline FH deletion in hereditary leiomyomatosis and renal cell carcinoma. Genes Chromosomes Cancer 56 (6): 484-492, 2017.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Forde C, Lim DHK, Alwan Y, et al.: Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur Urol Oncol 3 (6): 764-772, 2020.
Shuch B, Li S, Risch H, et al.: Estimation of the carrier frequency of fumarate hydratase alterations and implications for kidney cancer risk in hereditary leiomyomatosis and renal cancer. Cancer 126 (16): 3657-3666, 2020.
Lu E, Hatchell KE, Nielsen SM, et al.: Fumarate hydratase variant prevalence and manifestations among individuals receiving germline testing. Cancer 128 (4): 675-684, 2022.
Alam NA, Olpin S, Leigh IM: Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol 153 (1): 11-7, 2005.
Pfaffenroth EC, Linehan WM: Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther 8 (6): 779-90, 2008.
Grubb RL, Franks ME, Toro J, et al.: Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177 (6): 2074-9; discussion 2079-80, 2007.
Abou Alaiwi S, Nassar AH, Adib E, et al.: Trans-ethnic variation in germline variants of patients with renal cell carcinoma. Cell Rep 34 (13): 108926, 2021.
Zavoshi S, Lu E, Boutros PC, et al.: Fumarate Hydratase Variants and Their Association With Paraganglioma/Pheochromocytoma. Urology 176: 106-114, 2023.
Alam NA, Barclay E, Rowan AJ, et al.: Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141 (2): 199-206, 2005.
Coughlin EM, Christensen E, Kunz PL, et al.: Molecular analysis and prenatal diagnosis of human fumarase deficiency. Mol Genet Metab 63 (4): 254-62, 1998.
Bourgeron T, Chretien D, Poggi-Bach J, et al.: Mutation of the fumarase gene in two siblings with progressive encephalopathy and fumarase deficiency. J Clin Invest 93 (6): 2514-8, 1994.
Badeloe S, Frank J: Clinical and molecular genetic aspects of hereditary multiple cutaneous leiomyomatosis. Eur J Dermatol 19 (6): 545-51, 2009 Nov-Dec.
Kamihara J, Horton C, Tian Y, et al.: Different Fumarate Hydratase Gene Variants Are Associated With Distinct Cancer Phenotypes. JCO Precis Oncol 5: 1568-1578, 2021.
Kiuru M, Lehtonen R, Arola J, et al.: Few FH mutations in sporadic counterparts of tumor types observed in hereditary leiomyomatosis and renal cell cancer families. Cancer Res 62 (16): 4554-7, 2002.
Lehtonen R, Kiuru M, Vanharanta S, et al.: Biallelic inactivation of fumarate hydratase (FH) occurs in nonsyndromic uterine leiomyomas but is rare in other tumors. Am J Pathol 164 (1): 17-22, 2004.
Linehan WM, Spellman PT, Ricketts CJ, et al.: Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med 374 (2): 135-45, 2016.
Aspectos de la biología molecular
Actualmente se están investigando los mecanismos por los que las alteraciones en FH conducen a la leiomiomatosis hereditaria y cáncer de células renales (HLRCC). Se ha demostrado que la inactivación bialélica de FH produce pérdida de la fosforilación oxidativa y dependencia de la glucólisis aeróbica para satisfacer los requisitos energéticos celulares. La interrupción del ciclo de Krebs debido a la disminución o ausencia de actividad de la fumarato–hidratasa aumenta la concentración intracelular de fumarato. Este aumento inhibe la actividad de las prolilhidroxilasas del factor inducible por hipoxia (HIF), lo que conlleva acumulación de HIF-α.[1,2] Las variantes inactivadoras de FH también producen especies reactivas de oxígeno, lo que contribuyen aún más a estabilizar el HIF-α.[3] La activación de la vía del HIF produce un estado pseudohipóxico y un aumento regulado de un programa transcripcional que favorece el crecimiento tumoral rápido.[4] Además, la acumulación de fumarato activa la vía de la respuesta antioxidante, que permite la supervivencia de las células cancerosas en un entorno de estrés oxidativo. El fumarato, un electrófilo, es capaz de producir una modificación postranslacional de KEAP1 mediante succinación de los sulfhidrilos de cisteína,[5] de manera que libera la inhibición de KEAP1 sobre NRF2. La consecuente estabilización de NRF2 produce un aumento regulado de la transcripción de genes controlados por elementos de la respuesta antioxidante, como AKR1B10, lo que quizás contribuya con el proceso neoplásico.[6]
Referencias:
Isaacs JS, Jung YJ, Mole DR, et al.: HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8 (2): 143-53, 2005.
Pollard PJ, Brière JJ, Alam NA, et al.: Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14 (15): 2231-9, 2005.
Sudarshan S, Sourbier C, Kong HS, et al.: Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 29 (15): 4080-90, 2009.
Pollard P, Wortham N, Barclay E, et al.: Evidence of increased microvessel density and activation of the hypoxia pathway in tumours from the hereditary leiomyomatosis and renal cell cancer syndrome. J Pathol 205 (1): 41-9, 2005.
Alderson NL, Wang Y, Blatnik M, et al.: S-(2-Succinyl)cysteine: a novel chemical modification of tissue proteins by a Krebs cycle intermediate. Arch Biochem Biophys 450 (1): 1-8, 2006.
Ooi A, Wong JC, Petillo D, et al.: An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20 (4): 511-23, 2011.
Manifestaciones clínicas
Las características clínicas de la leiomiomatosis hereditaria y cáncer de células renales (HLRCC) son los leiomiomas cutáneos, los leiomiomas uterinos (fibromas) y el cáncer de células renales (RCC). Las personas afectadas presentan cualquiera de las siguientes manifestaciones: uno o más leiomiomas cutáneos o ausencia de lesiones cutáneas; RCC, que por lo general es solitario o ausencia de tumores renales; y leiomiomas uterinos. El fenotipo de la HLRCC es variable; la gravedad de la enfermedad exhibe variación significativa a nivel intrafamiliar e interfamiliar.[1,2,3]
Leiomiomas cutáneos
Los leiomiomas cutáneos son pápulas o nódulos firmes de color rosado o marrón rojizo. Estas lesiones por lo general aparecen en el tronco, las extremidades y, en ocasiones, en la cara. Se manifiestan a una mediana de edad de 25 años (intervalo de edad, 10–47 años) y suelen aumentar de tamaño y número con la edad. Son sensibles al tacto suave o al frío, y en ocasiones son dolorosas. La intensidad del dolor se correlaciona con la gravedad del compromiso cutáneo.[3] La presencia de varios leiomiomas cutáneos a menudo se relaciona con la HLRCC, y por lo tanto se justifica la evaluación genética. En una serie se notificó la presencia de leiomiomas cutáneos en 22 de 48 pacientes (46 %) que tenían una variante probablemente patogénica en FH.[4] En la misma serie se demostró que 18 de 19 personas (95 %) con leiomiomas múltiples tenían una variante patogénica en FH.[4] Por consiguiente, la presencia de un leiomioma solitario exige un análisis minucioso de la historia familiar de la persona. Para obtener más información, consultar las secciones Diagnóstico clínico y Diagnóstico diferencial.
Leiomiomas uterinos
Los leiomiomas uterinos en mujeres con HLRCC aparecen a una edad más temprana que en las mujeres de la población general. La edad de diagnóstico oscila entre 18 y 63 años (mediana de edad, 30 años). En una serie se notificaron leiomiomas uterinos en 18 de 29 mujeres (62 %) con una variante patogénica o probablemente patogénica de FH; la edad de las mujeres osciló entre 24 y 63 años.[4] Los leiomiomas uterinos suelen ser grandes y numerosos. La mayoría de las mujeres con HLRCC notifican menstruación irregular o abundante y dolor pélvico; por lo tanto, necesitan tratamiento de los leiomiomas a una edad más temprana que las mujeres de la población general. Las mujeres con HLRCC se someten a histerectomía o miomectomía por leiomiomas uterinos sintomáticos a una edad más temprana (<30 años) que las mujeres de la población general (mediana de edad, 45 años).[3,5,6,7]
Cáncer de células renales
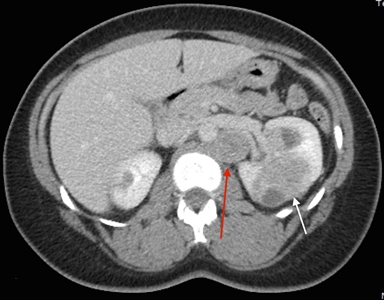
Los síntomas del cáncer de células renales (RCC) son hematuria, dolor lumbar y masa palpable. Sin embargo, muchas personas con RCC son asintomáticas. Además, no todas las personas con HLRCC presentarán RCC, ni será parte del cuadro clínico inicial. La mayoría de los RCC son unilaterales y solitarios; pero en algunas personas, son multifocales. Todavía no se ha determinado el incidencia exacta del RCC en personas afectadas, las estimaciones realizadas por diferentes grupos oscilan mucho (1–60 %).[1,3,8] La incidencia varía en función del lugar donde se realizó el estudio, las pautas de derivación de grupos individuales, y los exámenes de detección para el RCC a los que se someten los participantes. En los estudios del Instituto Nacional del Cáncer (NCI), se identificó RCC en el 32 % de las familias evaluadas.[1,3] La mediana de edad en el momento de la detección del RCC fue de 37 años,[9] aunque se notificaron algunos casos en edades tan tempranas como 10 años.[10] En otra serie grande de 135 pacientes se calculó que el riesgo de por vida fue de un 20,8 % a los 85 años de edad.[11] A diferencia de otros síndromes hereditarios de RCC, el RCC asociado a la HLRCC es muy maligno,[12,13] con un grado nuclear de Fuhrman de 3 o 4 en muchos casos; 9 de 13 personas murieron por enfermedad metastásica en el transcurso de 5 años del diagnóstico.[3] En la Figura 1 se observan varias lesiones de RCC en un paciente con HLRCC.
Leiomiosarcomas uterinos
No está claro si las mujeres con HLRCC tienen un riesgo más alto de leiomiosarcomas uterinos que las mujeres de edad similar en la población general. En la descripción original de la HLRCC, se notificó que 2 de 11 mujeres con leiomiomas uterinos también presentaban un leiomiosarcoma uterino, un tipo de cáncer que a veces es clínicamente muy maligno si no se detecta y se trata en estadio temprano.[2] Hasta el momento, se han notificado variantes patogénicas germinales de FH en 6 mujeres con leiomiosarcoma uterino.[14,15] Parece que la mayoría de las familias que obtienen un resultado positivo para una variante patogénica de FH no exhiben una predisposición elevada al cáncer uterino, pero unas pocas personas y familias sí tienen un riesgo alto. En los estudios de América del Norte, no se han notificado leiomiosarcomas uterinos en personas o familias con HLRCC.[3] Por lo tanto, el riesgo de leiomiosarcoma uterino en mujeres con HLRCC es incierto. Esta es una interrogante que debe resolverse con urgencia.
Otras manifestaciones
Se notificaron 4 casos con alteración en FH que presentaron cáncer de mama, 1 caso de cáncer de vejiga y 1 caso de enfermedad adrenocortical macronodular bilateral con síndrome de Cushing. En una serie del NCI se encontró que 20 de 255 pacientes (7,8 %) con HLRCC tenían nódulos suprarrenales, algunos que no parecían ser adenomas según sus características imaginológicas. Se extirparon muchas de estas lesiones porque eran ávidas de fluorodesoxiglucosa. En todas ellas se observó hiperplasia suprarrenal micronodular y macronodular, lo que indica que los nódulos adrenales quizás sean otra manifestación de la HLRCC.[16] Queda por resolver si estas manifestaciones son verdaderas características del fenotipo de la HLRCC.[6,14,17] De manera similar, se han notificado feocromocitomas en las glándulas suprarrenales.[8,18] No obstante, en una de las cohortes más grandes del Reino Unido, no se notificaron feocromocitomas en personas con HLRCC. Por lo tanto, no se recomienda la vigilancia de los feocromocitomas a menos que la evidencia nueva indique que este tumor hace parte del fenotipo de la HLRCC.[11]
Referencias:
Wei MH, Toure O, Glenn GM, et al.: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43 (1): 18-27, 2006.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Bhola PT, Gilpin C, Smith A, et al.: A retrospective review of 48 individuals, including 12 families, molecularly diagnosed with hereditary leiomyomatosis and renal cell cancer (HLRCC). Fam Cancer 17 (4): 615-620, 2018.
Farquhar CM, Steiner CA: Hysterectomy rates in the United States 1990-1997. Obstet Gynecol 99 (2): 229-34, 2002.
Alam NA, Barclay E, Rowan AJ, et al.: Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141 (2): 199-206, 2005.
Stewart L, Glenn GM, Stratton P, et al.: Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch Dermatol 144 (12): 1584-92, 2008.
Muller M, Ferlicot S, Guillaud-Bataille M, et al.: Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers. Clin Genet 92 (6): 606-615, 2017.
Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
Wong MH, Tan CS, Lee SC, et al.: Potential genetic anticipation in hereditary leiomyomatosis-renal cell cancer (HLRCC). Fam Cancer 13 (2): 281-9, 2014.
Forde C, Lim DHK, Alwan Y, et al.: Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur Urol Oncol 3 (6): 764-772, 2020.
Grubb RL, Franks ME, Toro J, et al.: Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177 (6): 2074-9; discussion 2079-80, 2007.
Linehan WM, Bratslavsky G, Pinto PA, et al.: Molecular diagnosis and therapy of kidney cancer. Annu Rev Med 61: 329-43, 2010.
Lehtonen HJ, Kiuru M, Ylisaukko-Oja SK, et al.: Increased risk of cancer in patients with fumarate hydratase germline mutation. J Med Genet 43 (6): 523-6, 2006.
Ylisaukko-oja SK, Kiuru M, Lehtonen HJ, et al.: Analysis of fumarate hydratase mutations in a population-based series of early onset uterine leiomyosarcoma patients. Int J Cancer 119 (2): 283-7, 2006.
Shuch B, Ricketts CJ, Vocke CD, et al.: Adrenal nodular hyperplasia in hereditary leiomyomatosis and renal cell cancer. J Urol 189 (2): 430-5, 2013.
Matyakhina L, Freedman RJ, Bourdeau I, et al.: Hereditary leiomyomatosis associated with bilateral, massive, macronodular adrenocortical disease and atypical cushing syndrome: a clinical and molecular genetic investigation. J Clin Endocrinol Metab 90 (6): 3773-9, 2005.
Clark GR, Sciacovelli M, Gaude E, et al.: Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 99 (10): E2046-50, 2014.
Características histopatológicas
Leiomiomas cutáneos
Se cree que los leiomiomas cutáneos surgen del músculo erector del pelo de los folículos pilosos. Desde el punto de vista histológico, son tumores dérmicos que no afectan la epidermis. Desde el punto de vista morfológico, estos tumores están formados por fibras de músculo liso entrelazadas con fibras de colágeno.[1]
Leiomiomas uterinos
En una revisión de la experiencia del Instituto Nacional del cáncer sobre leiomiomas uterinos asociados a la leiomiomatosis hereditaria y cáncer de células renales (HLRCC), se notificó que la mayoría de estos casos eran tumores fasciculares bien circunscritos, y algunos pocos casos exhibían un aumento de celularidad y atipia. La característica distintiva de estos casos fue similar a la observada en el cáncer de riñón de la HLRCC: presencia de nucléolos prominentes que se tiñen de color naranja y están rodeados por un halo perinuclear. Si bien algunos casos exhibieron características atípicas, no se notificaron lesiones con necrosis tumoral ni mitosis atípicas indicadoras de neoplasia maligna o leiomiosarcoma.[2]
Cáncer de células renales
El cáncer de células renales (RCC) propio de la HLRCC exhibe características histológicas únicas, como la presencia de células con abundante citoplasma anfófilo y núcleos grandes con nucléolos eosinofílicos grandes similares a cuerpos de inclusión. En la descripción original, estas características citológicas se atribuyeron a los tumores papilares de tipo 2.[3] No obstante, en los estudios iniciales se notificó que la HLRCC se relaciona con varios tumores renales que abarcan desde el tumor papilar de tipo 2 y el tumor tubulopapilar hasta el carcinoma de conducto colector.[4,5] El RCC de la HLRCC quizás corresponda a una nueva entidad patológica renal o un tipo propio de la HLRCC. En 2 estudios se describió la variedad de tipos morfológicos del RCC en el síndrome de HLRCC después de obtener análisis histológicos de 40 casos de RCC en 38 pacientes con variantes patogénicas germinales de FH e historia familiar compatible con HLRCC.[5,6] Se observaron varias configuraciones histológicas: quística, tubulopapilar, túbulo-sólida y mezcladas.[5,6]
Referencias:
Fearfield LA, Smith JR, Bunker CB, et al.: Association of multiple familial cutaneous leiomyoma with a uterine symplastic leiomyoma. Clin Exp Dermatol 25 (1): 44-7, 2000.
Sanz-Ortega J, Vocke C, Stratton P, et al.: Morphologic and molecular characteristics of uterine leiomyomas in hereditary leiomyomatosis and renal cancer (HLRCC) syndrome. Am J Surg Pathol 37 (1): 74-80, 2013.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Wei MH, Toure O, Glenn GM, et al.: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43 (1): 18-27, 2006.
Merino MJ, Torres-Cabala C, Pinto P, et al.: The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31 (10): 1578-85, 2007.
Merino MJ, Torres-Cabala CA, Zbar B, et al.: Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC): Clinical histopathological and molecular features of the first American families described. [Abstract] Mod Pathol 16 (Suppl): A-739, 162A, 2003.
Atención médica
Diagnóstico y pruebas
Las pruebas genéticas para el gen FH están disponibles en el ámbito clínico y se hacen en laboratorios certificados según las Enmiendas para el Mejoramiento de Laboratorios Clínicos (CLIA). En la actualidad, el gen FH es el único que se sabe que está relacionado con la leiomiomatosis hereditaria y carcinoma de células renales (HLRCC). La mayoría de los pacientes con HLRCC tienen una variante patogénica germinal de FH.
Debido a que el análisis genético de la HLRCC es complejo, cualquier interpretación de un informe de una variante de significado incierto debe estar a cargo del genetista que conduce la consulta en el ámbito oncológico, idealmente en un centro con bastante experiencia en esta enfermedad.
Diagnóstico clínico
No hay consenso actual sobre los criterios diagnósticos para la HLRCC.[1] Las directrices vigentes se crearon durante la era de clasificación histológica cuando el carcinoma de células renales (RCC) papilar con deficiencia de FH se clasificaba como RCC papilar de tipo 2. Las directrices quizás requieran actualización de acuerdo con el cambio en la clasificación del RCC, la disponibilidad de pruebas de tinción inmunohistoquímica para identificar la pérdida de FH y la presencia del marcador 2SC en las manifestaciones tisulares.
Algunos expertos sugieren que para determinar el diagnóstico clínico dermatológico de la HLRCC se requiere la presencia de una de las siguientes manifestaciones:[2]
Leiomiomas cutáneos múltiples, por lo menos uno de ellos confirmado por análisis histológico.
Un solo leiomioma en presencia de historia familiar compatible con la HLRCC.
De manera más reciente se han planteado criterios más amplios para determinar el diagnóstico, que a menudo son utilizados por expertos en el ámbito clínico. Los criterios sugeridos incluyen las manifestaciones dermatológicas como los leiomiomas cutáneos múltiples (confirmados por análisis histológico). Además, deben estar presentes por lo menos dos de las siguientes manifestaciones:[3,4]
Tratamiento quirúrgico de leiomiomas uterinos sintomáticos antes de los 40 años de edad.
Diagnóstico de RCC papilar de tipo 2 antes de los 40 años (que ahora se conoce como RCC papilar con deficiencia de FH).
Un familiar de primer grado que cumple uno de estos criterios.
Debido a que el RCC con deficiencia de FH se parece al RCC de conducto colector, la presencia de RCC con deficiencia de FH antes de los 40 años corresponde a otro criterio.[5] Los pacientes con tumores aparentemente esporádicos que no tienen una historia familiar compatible y que presentan un solo leiomioma cutáneo confirmado por análisis histológico, a veces obtienen un resultado positivo para una variante patogénica germinal de FH. Si bien se desconoce el porcentaje de variantes patogénicas germinales en estas poblaciones de pacientes, es posible que muchos centros deriven a pacientes con un solo leiomioma cutáneo para obtener asesoramiento y pruebas genéticas, independientemente de la historia familiar.[6]
Diagnóstico diferencial
Lesiones cutáneas
Los leiomiomas cutáneos son raros. La detección de lesiones múltiples es específica de la HLRCC. Debido a que el aspecto clínico de los leiomiomas es similar al aspecto de varias lesiones cutáneas, se exige el diagnóstico histológico para comprobar de manera objetiva la naturaleza de la lesión.
Leiomiomas uterinos
El leiomioma uterino es el tumor pélvico benigno más común en mujeres de la población general. La mayoría de los leiomiomas uterinos son esporádicos y no sindrómicos.[7]
Cáncer de células renales
Los indicios diagnósticos de la HLRCC se basan en la presencia de varias características fenotípicas en diferentes órganos (piel, útero y riñón). Es posible que una o más de estas características se presenten en el paciente o en uno o más de los familiares biológicos afectados.
Aunque el RCC familiar se vincula con una afección patológica renal bastante específica, la rareza de estos síndromes hace que pocos patólogos logren obtener experiencia suficiente como para reconocer sus características histológicas.
El diagnóstico diferencial abarca otros síndromes de RCC familiar raros que presentan características patológicas renales específicas, como los siguientes:
Carcinoma renal papilar hereditario (HPRC). En este caso hay predisposición al RCC papilar (antes conocido como RCC papilar de tipo 1). La herencia es autosómica dominante.[8]
Síndrome de Birt-Hogg-Dubé (BHD). Una variedad de tumores renales que abarca el oncocitoma renal (benigno), el cáncer de células renales cromófobo (maligno), y una combinación de ambos tipos celulares (llamado tumor híbrido oncocítico).[9] Las personas con BDH a veces presentan fibrofoliculomas o tricodiscomas cutáneos, quistes pulmonares múltiples y neumotórax espontáneo. La herencia es autosómica dominante.[10,11]
Pruebas genéticas
Las pruebas genéticas se usan en el ámbito clínico para confirmar el diagnóstico de las personas en riesgo. Se recomienda que se ofrezca asesoramiento genético previo y posterior a las pruebas a todas las personas que estén pensando en someterse a pruebas de variantes patogénicas germinales.[12] Los laboratorios para las pruebas genéticas deben tener la certificación según las CLIAS con el fin de que los resultados sean útiles para la toma de decisiones clínicas.[13]
Estrategias para las pruebas
Se indica el uso de una prueba genética para identificar una variante patogénica germinal de FH en todas las personas con HLRCC y en aquellas en las que se sospeche esta enfermedad, sin importar la historia familiar. Esto incluye a las personas con leiomiomas cutáneos, como se describe en la sección Diagnóstico clínico en este resumen, y las personas con tumores renales que exhiben características histológicas compatibles con la HLRCC.[4,14,15] Para obtener más información, consultar la sección Características histopatológicas.
Riesgo en familiares
La HLRCC se hereda de manera autosómica dominante.[16] Si uno de los progenitores de un probando está afectado y tiene manifestaciones clínicas o una variante causal de la enfermedad, cada hermano o hermana del probando presenta un riesgo del 50 % de haber heredado la variante patogénica. Cada hijo o hija de una persona con HLRCC tiene una probabilidad del 50 % de heredar la variante patogénica. La intensidad de la gravedad clínica no es predecible. Hay pruebas genéticas prenatales disponibles en laboratorios que ofrecen pruebas prenatales personalizadas para familias en quienes que se ha identificado una variante patogénica en un miembro de la familia afectado.
Progenitores de un probando
Algunas personas con diagnóstico de HLRCC tienen una historia familiar en la que solo un progenitor está afectado, mientras que en otros casos ambos progenitores se clasifican como no afectados, lo que indica que algunas personas presentan la HLRCC como resultado de una variante patogénica de novo o un mosaicismo parental.
Se desconoce la proporción de casos causados por variantes patogénicas de novo, ya que no se ha investigado de manera sistemática la presencia de manifestaciones sutiles en los progenitores, y no todos los progenitores no afectados se someten a pruebas de FH.
La evaluación de los progenitores de un probando con sospecha de una variante patogénica de novo quizás incluya pruebas genéticas si en el probando se identificó la variante causal de la enfermedad en FH.
Aunque algunas personas con diagnóstico de HLRCC tengan un progenitor afectado, la historia familiar quizás de la impresión de ser negativa porque la información es limitada, no se reconoció el trastorno en los familiares, el progenitor afectado murió antes de la aparición de los síntomas relacionados con el síndrome, o se produjo un inicio tardío de la enfermedad en el progenitor afectado.[17]
Hermanos y hermanas del probando
El riesgo para los hermanos o hermanas del probando depende del estado genético de los progenitores del probando.
Si uno de los progenitores del probando está afectado y exhibe manifestaciones clínicas o tiene una variante causal de enfermedad, cada uno de los hermanos o hermanas del probando tiene un 50 % de riesgo de heredar la variante.
Si la variante causal de enfermedad no se detecta en el DNA de ninguno de los progenitores, el riesgo para los hermanos o hermanas es bajo. Sin embargo, el riesgo será más alto que el riesgo en la población general por la posibilidad de un mosaicismo en la línea germinal.
Pruebas de familiares en riesgo
El uso de pruebas genéticas para la identificación temprana de familiares en riesgo mejora la certeza diagnóstica. Asimismo, reduce el número de procedimientos de detección innecesarios, estresantes y costosos en los miembros en riesgo que no heredaron la variante causal de enfermedad de su familia.[13,18,19]
El reconocimiento temprano de las manifestaciones clínicas quizás permita una intervención oportuna, que en teoría, podría mejorar el desenlace. Por lo tanto, es razonable establecer una vigilancia clínica de los familiares asintomáticos en riesgo con el fin de lograr la detección temprana del RCC, pero se necesitan otros datos objetivos sobre la repercusión de los exámenes de detección en la mortalidad vinculada con este síndrome.
Aspectos del asesoramiento genético
Predicción del fenotipo en personas que heredaron una variante patogénica
No es posible predecir si aparecerán síntomas propios de la HLRCC, y si aparecen, no se sabe la edad de inicio, el tipo, la gravedad ni las características clínicas que presentarán las personas con una variante patogénica. En una descripción profunda de las características clínicas y genéticas analizadas en 21 familias nuevas, los fenotipos fueron muy diversos desde el punto de vista clínico, y no se encontraron correlaciones entre genotipo y fenotipo. [20]
Cuando ninguno de los progenitores de un probando con una afección autosómica dominante presenta la variante causal de la enfermedad o cuando no hay indicios clínicos de la afección, es probable que el probando tenga una variante patogénica de novo. Sin embargo, entre las posibles explicaciones no médicas está la posibilidad de ausencia de paternidad biológica o una adopción no divulgada. Es apropiado obtener pruebas genéticas en los familiares en riesgo para identificar la necesidad de vigilancia clínica continua de por vida. La interpretación del resultado de la prueba de la variante patogénica es más exacta cuando se ha identificado una variante causal de la enfermedad en un familiar afectado. Se recomienda que los familiares que tienen una variante causal de la enfermedad se sometan a vigilancia periódica de por vida. Mientras tanto, los familiares y los descendientes que no heredaron la variante patogénica tienen un riesgo de RCC similar al riesgo de la población general; no se recomienda un seguimiento especial para estas personas.
La detección temprana de las personas en riesgo afecta su atención médica
Programar exámenes de detección para identificar manifestaciones tempranas de la HLRCC es un aspecto importante de la atención clínica de las personas afectadas. Aunque no hay estudios prospectivos en los que se comparen las prácticas de detección específicas para el cáncer renal, la naturaleza maligna del RCC relacionado con la HLRCC [3] justifica dirigir los esfuerzos a la identificación temprana del cáncer. Cuando los tumores son pequeños y localizados, es posible que una nefrectomía parcial sea una opción factible; sin embargo, la naturaleza infiltrativa de estos tumores ha llevado a algunos grupos a sugerir que se debe obtener un margen amplio para lograr una resección completa.[21]
Los fibromas uterinos a menudo causan síntomas importantes vinculados con hemorragias y presencia de una masa grande; aunque los fibromas pequeños a veces son asintomáticos. Dado que los fibromas de la HLRCC a veces exigen una histerectomía y la pérdida de la capacidad reproductiva en las jóvenes afectadas, el objetivo del examen de detección en las mujeres interesadas es conservar la fertilidad y limitar algunas de estas complicaciones irreversibles. Aunque no hay recomendaciones específicas para el tratamiento de los fibromas propios de la HLRCC, varios abordajes han demostrado ser eficaces en casos de fibromas esporádicos. Estas estrategias incluyen el uso de terapia hormonal, analgésicos, procedimientos percutáneos, endovasculares y quirúrgicos. La derivación temprana a un especialista en fertilidad a veces es útil para facilitar la planificación familiar.
Vigilancia
Se ha sugerido que las personas en quienes se sospecha o se confirmó el diagnóstico de la HLRCC, aquellas con variantes patogénicas heterocigotas de FH, independientemente de las manifestaciones clínicas, y los familiares en riesgo que no se han sometido a pruebas genéticas, continúen con vigilancia regular a cargo de médicos familiarizados con las manifestaciones clínicas de la HLRCC, como se describe a continuación.
Vigilancia cutánea. Se han publicado recomendaciones de exámenes regulares de la piel, pero no hay consenso en cuanto a la frecuencia de estos exámenes, y las recomendaciones no se han validado de manera prospectiva.
Vigilancia uterina. En las mujeres con útero intacto, se recomienda una consulta ginecológica cada año, acompañada de imágenes periódicas. Los métodos abarcan las imágenes por resonancia magnética (IRM) de la pelvis o una ecografía para evaluar la gravedad de los leiomiomas uterinos y buscar cambios que indiquen la aparición de un leiomiosarcoma.[6,7,16,22]
Vigilancia de paragangliomas o feocromocitomas. Si bien la vigilancia de paragangliomas o feocromocitomas no se incluye en las pautas recomendadas para la HLRCC, en un estudio se indicó que para las personas con variantes de FH relacionadas con riesgo de paragangliomas o feocromocitomas (es decir, p.Thr234Ala, p.Ala273Thr y p.Ala194Thr) se debería considerar una vigilancia similar a la recomendada para portadores de SDHx.[23] Este régimen de vigilancia incluye exámenes de detección bioquímicos anuales (metanefrinas en orina o plasma), examen físico minucioso, en especial en las áreas del cuello y el abdomen, e imágenes transversales de todo el cuerpo.
Vigilancia renal. En vista de la naturaleza maligna de esta enfermedad, se justifica obtener cada año imágenes de tomografía computarizada (TC) con contraste o IRM con gadolinio, incluso si durante la evaluación inicial (referencia) los riñones son normales. En esta población se justifica prestar atención especial a las imágenes porque incluso hallazgos sutiles, como un quiste complejo, a veces indican una neoplasia maligna agresiva. Sin embargo, no está clara la edad de inicio de los exámenes de detección porque la HLRCC se ha descrito en niños de tan solo 10 años. La HLRCC Family Alliance recomienda obtener imágenes anuales desde los 8 años en niños en riesgo de HLRCC o en quienes ya se diagnosticó la HLRCC.[1] La IRM tiene la ventaja de evitar la exposición a la radiación y por este motivo se prefiere a la TC para la vigilancia de por vida en los pacientes con HLRCC.
Cualquier lesión renal sospechosa (quistes indeterminados, dudosos o complejos) identificada en un examen previo debe seguirse de cerca con imágenes periódicas, preferiblemente con la misma modalidad para permitir comparaciones. El uso de ecografía renal a veces es útil para la caracterización de lesiones quísticas identificadas en las imágenes transversales. Se debe advertir que la ecografía por sí sola nunca es suficiente. Los tumores renales los debe evaluar un médico familiarizado con el cáncer renal propio de la HLRCC.[10,24]
Debido al rápido crecimiento de estos tumores, se justifica la vigilancia regular usando un umbral bajo para decidir la intervención quirúrgica temprana en el caso de lesiones renales sólidas. Esta estrategia difiere de la descrita para otros síndromes hereditarios de RCC, en donde el comportamiento del tumor por lo general es de crecimiento lento, y para quienes la observación es una opción viable.[10,24,25]
Nivel de evidencia (vigilancia cutánea): 5
Nivel de evidencia (vigilancia uterina): 4
Nivel de evidencia (vigilancia renal): 4
Tratamiento de las manifestaciones
Lesiones cutáneas
El especialista en dermatología es el médico idóneo para evaluar los leiomiomas cutáneos. Por lo general, los leiomiomas cutáneos asintomáticos no requieren tratamiento. El tratamiento de los leiomiomas cutáneos sintomáticos a veces es difícil si la enfermedad del paciente es difusa y de distribución amplia. La extirpación es posible en el caso de lesiones solitarias dolorosas. El tratamiento de las lesiones se hace con crioablación o láser. Se ha notificado que varios fármacos, como los bloqueantes de los canales de calcio, los alfabloqueantes, la nitroglicerina, los antidepresivos y los antiepilépticos, reducen el dolor de los leiomiomas.[26] En un ensayo clínico aleatorizado (09-C-0072 [NCT00971620]) se observó que la inyección intralesional de toxina botulínica de tipo A (Botox) quizás mejore la calidad de vida.[27]
Nivel de evidencia: 5
Leiomiomas uterinos
El especialista en ginecología es el médico idóneo para evaluar los leiomiomas uterinos. Los leiomiomas propios de la HLRCC se tratan de la misma manera que los leiomiomas esporádicos. Sin embargo, debido a la multiplicidad, el tamaño y el posible crecimiento rápido de los leiomiomas uterinos propios de la HLRCC, es posible que la mayoría de las mujeres necesiten intervención médica o quirúrgica a una edad más temprana y con más frecuencia de lo que se esperaría en la población general. El tratamiento médico (en la actualidad con agonistas de la hormona liberadora de gonadotropina, antihormonales y analgésicos) inicial de los leiomiomas uterinos sirve para disminuir el tamaño, preparar la lesión para la extirpación quirúrgica y brindar alivio temporal del dolor. Cuando las mujeres desean conservar la fertilidad, es posible usar una miomectomía para extirpar los leiomiomas y conservar el útero. El histerectomía solo se debe realizar cuando sea necesario.[6,7]
Nivel de evidencia: 4
Cáncer de células renales
Es prudente establecer esfuerzos dirigidos a la detección temprana del RCC propio de la HLRCC, por su grado de malignidad biológica. Sin embargo, en los estudios vigentes no se ha demostrado que la detección temprana se relacione de manera clara con una mejora de la supervivencia. Se recomienda la extirpación quirúrgica de estas neoplasias malignas cuando aparezca el primer signo de la enfermedad, a diferencia del tratamiento de otros síndromes de cáncer hereditario. La propensión al compromiso ganglionar, incluso con tumores renales pequeños, quizás exija la disección de ganglios linfáticos para obtener una estadificación adecuada.[21] La nefrectomía radical o la nefrectomía parcial con márgenes amplios se debe considerar en personas que tienen masas renales detectables, incluso tumores pequeños que miden menos de 1 cm.[10,24,25]
Nivel de evidencia: 4
Tratamientos en investigación
Se ha indicado que la sobreexpresión del factor inducible por hipoxia 1-α (HIF1-α) explica, en parte, la carcinogénesis de la HLRCC.[28,29] Por lo tanto, aunque todavía no están disponibles, los fármacos que se dirigen a HIF1-α podrían usarse para una terapia dirigida a los tumores propios de la HLRCC.
La pérdida de la fosforilación oxidativa como resultado de la inactivación bialélica de FH hace que los tumores de la HLRCC dependan casi por completo de la glucólisis aeróbica para cumplir con los requisitos celulares de trifosfato de adenosina y otros requisitos bioenergéticos. Por lo tanto, se está explorando el uso de la glucólisis aeróbica como un posible objetivo terapéutico.[30,31] En un estudio de fase II (10-C-0114 [NCT01130519]) se está evaluando el uso de la combinación de bevacizumab y erlotinib para el tratamiento de la HLRCC. Esto se basa en parte en la premisa de que esta combinación podría inhibir la administración eficaz de glucosa a las células tumorales.[32]
Otras investigaciones [33] en las que se evaluaron las consecuencias conocidas de la inactivación de la FH en el cáncer de riñón propio de la HLRCC se confirmó una expresión de concentraciones muy altas de la NAD(P)H quinina–deshidrogenasa 1 (NQO1) en los tumores renales de la HLRCC. La expresión en otros tipos de RCC hereditarios, como los carcinomas renales de células claras relacionados con la enfermedad de Von Hippel-Lindau y los carcinomas renales papilares relacionados con HPRC. La activación de una vía de respuesta al estrés oxidativo mediada por NRF2, un factor de transcripción que regula la transcripción de NQO1, quizás explique la sobreexpresión de NQO1 en estos tumores. El vandetanib, un inhibidor oral de VEGFR2 y EGFR que también es activo contra la cinasa Abl-1, tiene una actividad intensa in vitro contra células con deficiencia de FH, e induce la regresión de xenoinjertos derivados de la HLRCC en ratones. La actividad del vandetanib en este modelo está mediada, al menos en parte, por su capacidad para alterar la vía citoprotectora de respuesta al estrés oxidativo mediada por NRF2 gracias a un mecanismo dependiente de Abl. Además, la metformina, un activador de la proteína cinasa activada por 5'–AMP (AMPK) tuvo un efecto sinérgico con el vandetanib, in vitro y en ratones con xenoinjertos derivados de cáncer de riñón humano con deficiencia de FH.[34] Estos datos fueron el fundamento para un ensayo clínico (NCT02495103) en el que se evaluó la eficacia de esta combinación en pacientes con HLRCC y cáncer renal avanzado.
HLRCC Family Alliance: The HLRCC Handbook. Version 2.0. Boston, MA: HLRCC Family Alliance, 2013. Available online. Last accessed November 6, 2024.
Schmidt LS, Linehan WM: Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Renovasc Dis 7: 253-60, 2014.
Smit DL, Mensenkamp AR, Badeloe S, et al.: Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet 79 (1): 49-59, 2011.
Menko FH, Maher ER, Schmidt LS, et al.: Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 13 (4): 637-44, 2014.
Lehtonen HJ: Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 10 (2): 397-411, 2011.
Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Stewart L, Glenn GM, Stratton P, et al.: Association of germline mutations in the fumarate hydratase gene and uterine fibroids in women with hereditary leiomyomatosis and renal cell cancer. Arch Dermatol 144 (12): 1584-92, 2008.
Zbar B, Tory K, Merino M, et al.: Hereditary papillary renal cell carcinoma. J Urol 151 (3): 561-6, 1994.
Pavlovich CP, Walther MM, Eyler RA, et al.: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 26 (12): 1542-52, 2002.
Linehan WM, Bratslavsky G, Pinto PA, et al.: Molecular diagnosis and therapy of kidney cancer. Annu Rev Med 61: 329-43, 2010.
Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 12 (10): 558-69, 2015.
Riley BD, Culver JO, Skrzynia C, et al.: Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns 21 (2): 151-61, 2012.
Update: information about obtaining a CLIA certificate. Jt Comm Perspect 26 (12): 6-7, 2006.
Kopp RP, Stratton KL, Glogowski E, et al.: Utility of prospective pathologic evaluation to inform clinical genetic testing for hereditary leiomyomatosis and renal cell carcinoma. Cancer 123 (13): 2452-2458, 2017.
Merino MJ, Torres-Cabala C, Pinto P, et al.: The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 31 (10): 1578-85, 2007.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Refae MA, Wong N, Patenaude F, et al.: Hereditary leiomyomatosis and renal cell cancer: an unusual and aggressive form of hereditary renal carcinoma. Nat Clin Pract Oncol 4 (4): 256-61, 2007.
American Society of Clinical Oncology: American Society of Clinical Oncology policy statement update: genetic testing for cancer susceptibility. J Clin Oncol 21 (12): 2397-406, 2003.
Robson ME, Storm CD, Weitzel J, et al.: American Society of Clinical Oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 28 (5): 893-901, 2010.
Wei MH, Toure O, Glenn GM, et al.: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 43 (1): 18-27, 2006.
Shuch B, Singer EA, Bratslavsky G: The surgical approach to multifocal renal cancers: hereditary syndromes, ipsilateral multifocality, and bilateral tumors. Urol Clin North Am 39 (2): 133-48, v, 2012.
Patel VM, Handler MZ, Schwartz RA, et al.: Hereditary leiomyomatosis and renal cell cancer syndrome: An update and review. J Am Acad Dermatol 77 (1): 149-158, 2017.
Zavoshi S, Lu E, Boutros PC, et al.: Fumarate Hydratase Variants and Their Association With Paraganglioma/Pheochromocytoma. Urology 176: 106-114, 2023.
Grubb RL, Franks ME, Toro J, et al.: Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177 (6): 2074-9; discussion 2079-80, 2007.
Pfaffenroth EC, Linehan WM: Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther 8 (6): 779-90, 2008.
Ritzmann S, Hanneken S, Neumann NJ, et al.: Type 2 segmental manifestation of cutaneous leiomyomatosis in four unrelated women with additional uterine leiomyomas (Reed's Syndrome). Dermatology 212 (1): 84-7, 2006.
Naik HB, Steinberg SM, Middelton LA, et al.: Efficacy of Intralesional Botulinum Toxin A for Treatment of Painful Cutaneous Leiomyomas: A Randomized Clinical Trial. JAMA Dermatol 151 (10): 1096-102, 2015.
Isaacs JS, Jung YJ, Mole DR, et al.: HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8 (2): 143-53, 2005.
Pollard PJ, Brière JJ, Alam NA, et al.: Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14 (15): 2231-9, 2005.
Xie H, Valera VA, Merino MJ, et al.: LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther 8 (3): 626-35, 2009.
Yamasaki T, Tran TA, Oz OK, et al.: Exploring a glycolytic inhibitor for the treatment of an FH-deficient type-2 papillary RCC. Nat Rev Urol 8 (3): 165-71, 2011.
Linehan WM, Pinto PA, Srinivasan R, et al.: Identification of the genes for kidney cancer: opportunity for disease-specific targeted therapeutics. Clin Cancer Res 13 (2 Pt 2): 671s-679s, 2007.
Sourbier C, Ricketts CJ, Matsumoto S, et al.: Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 26 (6): 840-50, 2014.
Pronóstico
El pronóstico es bastante bueno para las manifestaciones cutáneas y uterinas de la leiomiomatosis hereditaria y cáncer de células renales (HLRCC). Cuando es necesario, el tratamiento local de las manifestaciones cutáneas y la histerectomía, cuando está indicada, permite abordar estos sitios de manera bastante eficaz y con consecuencias mínimas a largo plazo. Es probable que la incidencia de los leiomiosarcomas uterinos sea bastante baja y es improbable que afecte de manera importante la mediana de supervivencia a nivel de la cohorte. El cáncer de células renales (RCC) en el contexto de la HLRCC es una manifestación mucho más preocupante, y los pacientes con HLRCC que presentan RCC [1,2,3,4,5] tienen un riesgo alto de enfermedad metastásica.[6] El RCC metastásico de la HLRCC se caracteriza por una evolución clínica muy maligna. No hay cohortes de pacientes ni bases de datos lo suficientemente grandes que permitan obtener una estimación precisa de la supervivencia en esta población; sin embargo, las cohortes retrospectivas indican que este tipo de cáncer acarrea un desenlace más precario que otras formas convencionales de RCC.[7]
Referencias:
Shuch B, Vourganti S, Ricketts CJ, et al.: Defining early-onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol 32 (5): 431-7, 2014.
Launonen V, Vierimaa O, Kiuru M, et al.: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 98 (6): 3387-92, 2001.
Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Alam NA, Barclay E, Rowan AJ, et al.: Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed tumor syndrome. Arch Dermatol 141 (2): 199-206, 2005.
Forde C, Lim DHK, Alwan Y, et al.: Hereditary Leiomyomatosis and Renal Cell Cancer: Clinical, Molecular, and Screening Features in a Cohort of 185 Affected Individuals. Eur Urol Oncol 3 (6): 764-772, 2020.
Grubb RL, Franks ME, Toro J, et al.: Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol 177 (6): 2074-9; discussion 2079-80, 2007.
Chen F, Zhang Y, Şenbabaoğlu Y, et al.: Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Rep 14 (10): 2476-89, 2016.
Indicaciones futuras
En la atención de los pacientes con leiomiomatosis hereditaria y cáncer de células renales (HLRCC) hay dos necesidades insatisfechas principales, aparte de la disponibilidad de un tratamiento médico eficaz para la enfermedad metastásica. La primera es la capacidad de predecir la aparición del cáncer de células renales de manera que se haga una detección temprana con un grado más alto de precisión. El desarrollo de pruebas de diagnóstico en muestras de sangre o por imágenes que permitan una vigilancia rentable de los pacientes con HLRCC tendría un efecto favorable importante en los desenlaces de estas personas. La segunda necesidad insatisfecha importante es una determinación más exacta de las correlaciones entre el genotipo y el fenotipo según el tipo de lesión genética que se encuentra en el gen FH. Los polimorfismos en el gen FH que se han identificado de manera reciente a menudo se clasifican como de significado incierto, y determinar su importancia clínica requiere mucho trabajo. El diseño informático (in silico) de herramientas de predicción informáticas y su vinculación con grandes bases de datos y registros de pacientes ampliarían la comprensión de variantes específicas del gen FH.
Actualizaciones más recientes a este resumen (02 / 18 / 2025)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se añadió texto para indicar que las directrices vigentes se crearon durante la era de clasificación histológica cuando el carcinoma de células renales (RCC) papilar con deficiencia de FH se clasificaba como RCC papilar de tipo 2. Las directrices quizás requieran actualización de acuerdo con el cambio en la clasificación del RCC, la disponibilidad de pruebas de tinción inmunohistoquímica para identificar la pérdida de FH y la presencia de 2SC en las manifestaciones tisulares.
Se añadió texto sobre los criterios sugeridos para el diagnóstico de leiomiomatosis hereditaria y carcinoma de células renales (HLRCC).
Se revisó el texto para indicar que a que el RCC con deficiencia de FH se parece al RCC de conducto colector, la presencia de RCC con deficiencia de FH antes de los 40 años corresponde a otro criterio para el diagnóstico de HLRCC.
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre la genética de la leiomiomatosis hereditaria y cáncer de células renales. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre la genética del cáncer, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Leiomiomatosis hereditaria y cáncer de células renales son:
Alexandra Perez Lebensohn, MS, CGC (National Cancer Institute)
Brian Matthew Shuch, MD (UCLA Health)
Ramaprasad Srinivasan, MD, PhD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre la genética del cáncer emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
La información en estos resúmenes no se debe utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.