Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre el retinoblastoma
El retinoblastoma es un cáncer infantil que exige una cuidadosa integración de la atención multidisciplinaria. El objetivo del tratamiento del retinoblastoma es salvar la vida del paciente y conservar la visión útil. En los pacientes que presentan retinoblastoma extraocular, es probable que el tratamiento con quimioterapia sistémica y radioterapia sea curativo. Sin embargo, la enfermedad extraorbitaria exige quimioterapia intensiva y a veces incluye consolidación con dosis altas de quimioterapia y rescate de células madre hematopoyéticas autógenas con radioterapia o sin esta. Aunque la mayoría de los pacientes con metástasis sistémicas fuera del sistema nervioso central (SNC) se pueden curar, el pronóstico de los pacientes con enfermedad intracraneal es muy desalentador.
Incidencia
El retinoblastoma es un tumor infantil relativamente poco frecuente que se origina en la retina. Representa alrededor del 3 % de los cánceres en los niños menores de 15 años.
El retinoblastoma es un cáncer que afecta a niños muy pequeños. Dos tercios de todos los casos de retinoblastoma se diagnostican antes de los 2 años.[1] En consecuencia, si bien el cálculo de la incidencia anual en los Estados Unidos es de alrededor de 3 casos por millón de personas menores de 20 años, la incidencia anual ajustada por edad en niños de 0 a 4 años es de 18,4 casos por millón.[2]
Características anatómicas
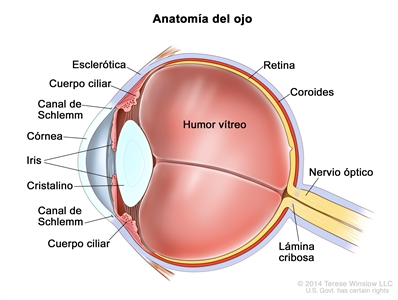
El retinoblastoma surge en la retina y suele crecer debajo de la retina y hacia la cavidad vítrea. El compromiso de las membranas oculares y el nervio óptico se produce en secuencia a medida que el tumor progresa.
La invasión focal de la coroides es frecuente, aunque la invasión masiva se produce en casos de enfermedad avanzada. Después de la invasión de la coroides, el tumor llega a la circulación sistémica y crea la posibilidad de metástasis. La progresión adicional a través de las membranas oculares conduce a la invasión de la esclerótica y la órbita. Los tumores que invaden la cámara anterior a veces acceden a la circulación sistémica a través del canal de Schlemm. La progresión por el nervio óptico y más allá de la lámina cribosa aumenta el riesgo de diseminación sistémica y al SNC (consultar la Figura 1).
Exámenes de detección
En informes de consenso de la American Association of Ophthalmic Oncologists and Pathologists y el American Association for Cancer Research Childhood Cancer Predisposition Workshop se describen las pautas de detección para niños en riesgo de presentar retinoblastoma.[3,4]
En los niños con antecedentes familiares de retinoblastoma se llevan a cabo exámenes de detección al comienzo de la vida mediante oftalmoscopia con anestesia general, a intervalos regulares. Los exámenes se realizan de acuerdo con el cálculo del riesgo absoluto a partir de la identificación de la variante de RB1 en la familia o la presencia de una variante de RB1 en el niño.[3,4]
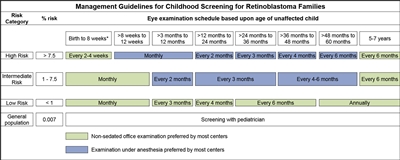
Los hijos de progenitores afectados se someten a una exploración ocular con dilatación pupilar y anestesia general tan pronto como sea posible durante el primer mes de vida, y también se efectúa una evaluación genética. Los lactantes que obtienen resultados positivos en las pruebas genéticas se someten a exámenes mensuales con anestesia. Los lactantes que no presentan la enfermedad continúan con exámenes mensuales durante el primer año de vida. La frecuencia de esos estudios se reduce de manera progresiva durante el segundo año y los siguientes. Los exámenes de detección en los niños con antecedentes familiares de retinoblastoma quizás mejoren el pronóstico, en términos de mantener la integridad del globo ocular gracias al empleo de tratamientos de conservación ocular menos radicales (consultar el Cuadro 1 y la Figura 2).[3,4]
Cuadro 1. Riesgo previo a la prueba para los familiares portadores del mismo alelo alterado deRB1que se identificó en el probandoa,b
Familiar del probando (cualquier sexo)
Riesgo previo a la prueba del alelo alterado (%)
Probando bilateral (100)
Probando unilateral (15)
a Reproducción deOphthalmology, Volumen 125, número 3, Alison H. Skalet, Dan S. Gombos, Brenda L. Gallie, Jonathan W. Kim, Carol L. Shields, Brian P. Marr, Sharon E. Plon, Patricia Chévez-Barrios, Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists, Páginas 453–458, Derechos de autor (2018), autorizado por Elsevier.
b Riesgo previo a la prueba para la variante deRB1en familiares de un niño afectado porretinoblastoma. El riesgo de presentar el alelo alterado deRB1se muestra como porcentaje en probandos unilaterales o bilaterales sin antecedentes familiares de retinoblastoma.
c Los familiares de tercer y cuarto grado de probandos unilaterales tienen un riesgo estimado de 0,003 y 0,001 %, respectivamente, que es más bajo que el riesgo de la población normal, de 0,007 % (1 en 15 000 nacidos vivos); por lo tanto, el riesgo se establece en 0,007 %.
Descendiente (lactante)
50
7,5
Progenitor
5
0,8
Hermano
2,5
0,4
Sobrino
1,3
0,2
Tío
0,1
0,007c
Primo de primer grado
0,05
0,007c
Población general
0,007
Es una práctica habitual que los padres y hermanos de ambos sexos de pacientes con retinoblastoma se sometan a exámenes de detección oftalmológicos con el fin de excluir una enfermedad familiar inadvertida. Sin embargo, cuando no se dispone de pruebas genéticas, el plan de exámenes de detección para un niño con un progenitor que tenga retinoblastoma unilateral no está bien definido.[5]
Cuadro clínico inicial
La edad en el momento del cuadro clínico inicial se correlaciona con lateralidad. Los pacientes con enfermedad bilateral exhiben manifestaciones a una edad más temprana, por lo general, en los primeros 12 meses de vida.
La mayoría de los pacientes presentan leucocoria, que en ocasiones se observa por primera vez después de tomar una fotografía con flash. El estrabismo es el segundo signo de presentación más frecuente y, por lo habitual, se correlaciona con compromiso macular. Los tumores intraoculares muy avanzados producen dolor, celulitis orbitaria, glaucoma o buftalmía.
A medida que el tumor progresa, los pacientes a veces presentan enfermedad orbitaria o metastásica. Se encuentran metástasis en los ganglios linfáticos preauriculares y laterocervicales, en el SNC o en forma sistémica (con frecuencia, en los huesos, la médula ósea y el hígado).
En los Estados Unidos, los niños hispanos y aquellos que viven en condiciones socioeconómicas precarias presentan una enfermedad más avanzada.[6]
Evaluación diagnóstica y estadificación
La evaluación diagnóstica del retinoblastoma incluye los siguientes procedimientos:
Examen ocular. El diagnóstico del retinoblastoma intraocular por lo general se establece sin confirmación patológica. Para inspeccionar la retina completa, es necesario una exploración con anestesia, una pupila dilatada al máximo e indentación escleral. Se debe documentar en detalle el número, la ubicación y el tamaño de los tumores; la presencia de desprendimiento de retina y de líquido subretiniano; y la presencia de diseminación subretiniana y vítrea.
Ecografía ocular e imágenes por resonancia magnética (IRM). La ecografía ocular bidimensional y las IRM son útiles para diferenciar el retinoblastoma de otras causas de leucocoria y para evaluar la diseminación fuera de la esclerótica y del ojo en niños con retinoblastoma intraocular avanzado. El realce del nervio óptico en la IRM no indica necesariamente que hay compromiso, de manera que estos hallazgos se deben interpretar con cautela.[7]
Quizás sea necesario evaluar la presencia de enfermedad metastásica en los pacientes en quienes se sospecha diseminación extraocular, por los hallazgos de las imágenes o un análisis patológico de riesgo alto en el ojo enucleado (es decir, invasión masiva de la coroides, compromiso de la esclerótica o del nervio óptico más allá de la lámina cribosa). Los pacientes que presentan estas características patológicas en el ojo enucleado tienen un riesgo alto de metástasis. En estos casos, a veces se hacen los siguientes procedimientos:[8]
Centellografía ósea.
Aspiración de médula ósea y biopsia.
Punción lumbar.
Características genéticas y genómicas del retinoblastoma
El retinoblastoma es un tumor que se presenta en forma hereditaria (25–30 %) y no hereditaria (70–75 %).
Retinoblastoma hereditario
El retinoblastoma hereditario se define por la presencia de una variante patogénica germinal del gen RB1. Esta variante patogénica germinal se hereda de un progenitor afectado (25 % de los casos), sucede en una célula germinal antes de la concepción o en el útero durante la embriogénesis temprana en pacientes con enfermedad esporádica (75 % de los casos). La presencia de antecedentes familiares de retinoblastoma, o enfermedad bilateral o multifocal puede indicar enfermedad hereditaria.
El retinoblastoma hereditario se manifiesta como enfermedad unilateral o bilateral. Es probable que la penetrancia de la variante de RB1 (lateralidad, edad en el momento del diagnóstico y número de tumores) dependa de modificadores genéticos simultáneos, como los polimorfismos de MDM2 y MDM4.[9,10] Se presume que todos los niños con enfermedad bilateral y cerca del 15 % de los pacientes con enfermedad unilateral tienen la forma hereditaria, a pesar de que solo el 25 % tienen un progenitor afectado. En una serie de 482 pacientes con retinoblastoma unilateral, se identificaron variantes patogénicas germinales en el 33 % de los lactantes menores de 12 meses, el 6 % de los niños de 12 a 24 meses y el 7 % de los niños de 24 a 39 meses. La mayor incidencia de retinoblastoma de la línea germinal se produjo en pacientes menores de 1 año, en comparación con los pacientes mayores de 1 año (oportunidad relativa, 2,96).[11][Nivel de evidencia C2]
En niños con retinoblastoma hereditario, el diagnóstico tiende a hacerse a una edad más temprana que en los niños con la forma no hereditaria de la enfermedad.[12]
Retinoblastoma no hereditario
El panorama actual de las características genómicas del retinoblastoma se orienta hacia las alteraciones en RB1 que producen inactivación bialélica.[13,14] Una causa poco frecuente de inactivación de RB1 es la cromotripsis, que es difícil de detectar con los métodos convencionales.[15]
Los cambios recurrentes en otros genes distintos de RB1 son poco frecuentes en el retinoblastoma, pero se producen. Las variantes o deleciones de BCOR y la amplificación de MYCN son los eventos que se notifican con más frecuencia.[13,14,15,16,17,18] En un estudio de 1068 casos de tumores unilaterales de retinoblastoma no familiar, se notificó que del 2 % al 3 % de los tumores carecían de pruebas de pérdida de RB1. Alrededor de la mitad de estos casos sin pérdida de RB1 exhibieron amplificación de MYCN.[14] Sin embargo, la amplificación de MYCN también se observa en los tumores de retinoblastoma que tienen alteraciones en RB1, lo que indica que la inactivación de RB1 por una variante o por la presencia de la proteína del retinoblastoma inactiva es un requisito para la presentación de un retinoblastoma, de manera independiente a la amplificación de MYCN.[19]
Asesoramiento genético
El asesoramiento genético es un componente fundamental de la atención de los pacientes con retinoblastoma y sus familias, con independencia del cuadro clínico inicial. El asesoramiento genético incluye una conversación sobre las principales formas de retinoblastoma, lo que ayuda a los padres a comprender las consecuencias genéticas de cada forma de retinoblastoma y permite calcular el riesgo de la enfermedad en la familia.[20] El asesoramiento también incluye orientación sobre los exámenes de detección adecuados para los progenitores y sus familiares, en especial, si hay aumento del riesgo de una segunda neoplasia maligna primaria.
Sin embargo, el asesoramiento genético no siempre es sencillo. Alrededor del 10 % de los niños con retinoblastoma exhiben mosaicismo genético somático, lo que dificulta este asesoramiento.[21] Los niños con mosaicismo alélico exhiben menos tumores, y es más probable que los tumores sean unilaterales.[22] Además, para una variante específica, el riesgo de retinoblastoma en los hermanos de ambos sexos varía en parte según el origen hereditario materno o paterno.[23] Para obtener más información, consultar Evaluación del riesgo de cáncer y asesoramiento genético.
Pruebas genéticas
Para determinar si un paciente con retinoblastoma tiene una variante germinal o somática del gen RB1, se examinan muestras de sangre o del tumor. Una vez que se identifica la variante genética del paciente, se procede a examinar a otros miembros de la familia para detectar directamente la variante mediante secuenciación dirigida.
Es posible realizar una evaluación genética completa del gen RB1 mediante un análisis de pasos múltiples, como los siguientes:[24]
Secuenciación de ADN para identificar variantes de los exones codificantes, en las regiones intrónicas circundantes y en las regiones promotoras.
Análisis de duplicaciones o deleciones.
Análisis de metilación de la región promotora de RB1 en ADN aislado del tumor.
En casos de mosaicismo somático o anomalías citogenéticas, es posible que no sea fácil detectar las variantes. Se deben utilizar técnicas más minuciosas, como el cariotipado, la hibridación fluorescente in situ y el análisis de metilación del promotor de RB1. A veces se descubre un mosaicismo de grado bajo mediante la secuenciación masiva (2500 veces) de un amplicón genómico de RB1 obtenido del ADN linfocítico.[25] Debido a que la causa del mosaicismo es una variante poscigótica, dicho resultado hace innecesaria la evaluación con anestesia general en los hermanos de ambos sexos. Las técnicas vigentes no detectarán algunas variantes mosaicas cuando se usan grados muy bajos de amplificación, tampoco detectan variantes ubicadas fuera de los exones codificantes de RB1 o en las regiones intrónicas circundantes, variantes en otros tejidos no linfocíticos ni reordenamientos grandes de tipo mosaico en RB1.[25] Al combinar las técnicas descritas antes es posible detectar una variante patogénica germinal en más del 90 % de los pacientes con retinoblastoma hereditario.[20,26,27]
La ausencia de variantes somáticas de RB1 detectables en cerca del 3 % de los casos de retinoblastoma unilateral no hereditario indica que quizás hay mecanismos subyacentes alternos que explican la formación de un retinoblastoma.[28] En la mitad de estos casos, se notificaron grados altos de amplificación de MYCN. Estos pacientes exhiben características histológicas de gran malignidad y una mediana de edad de 4 meses en el momento del diagnóstico.[14] No obstante, también se ha notificado la coexistencia de una amplificación de MYCN y variantes de RB1.[19] En otro pequeño subconjunto de tumores sin variantes somáticas de RB1 detectables, se encuentra inactivación del gen RB1 causada por cromotripsis.[15]
Vigilancia posterior al diagnóstico
Es posible que los niños con una variante patogénica germinal de RB1 continúen presentando tumores nuevos durante unos pocos años después del diagnóstico y tratamiento. Por esta razón, es necesario examinar con frecuencia a estos pacientes. Es una práctica habitual que se examinen cada 2 a 4 meses durante, por lo menos, 28 meses.[29] El intervalo entre exámenes se basa en la estabilidad de la enfermedad y la edad del niño (es decir, consultas menos frecuentes a medida que el niño crece).
Una proporción de los niños que presentan retinoblastoma unilateral, con el tiempo, presentarán la enfermedad en el ojo opuesto. Por este motivo, se realizan exámenes periódicos del ojo no afectado hasta que se determine el estado en la línea germinal del gen RB1.
Debido al pronóstico precario de los pacientes con retinoblastoma trilateral, es una práctica habitual realizar exámenes de detección con neuroimágenes hasta los 5 años de edad durante la vigilancia de los niños con formas hereditarias de la enfermedad. Para obtener más información, consultar la sección Retinoblastoma trilateral.
Causas de la mortalidad relacionada con el retinoblastoma
Si bien el retinoblastoma es una enfermedad muy curable, el reto es conservar la vida y prevenir la pérdida ocular, la ceguera y otros efectos graves del tratamiento que reducen la longevidad o la calidad de vida del paciente. Con las mejoras en el diagnóstico y el tratamiento del retinoblastoma en las últimas décadas, el retinoblastoma metastásico se observa con menor frecuencia en los Estados Unidos y otras naciones desarrolladas. Por este motivo, otras causas de mortalidad relacionada con el retinoblastoma, como el retinoblastoma trilateral y las neoplasias subsiguientes (NS), han cobrado mayor importancia durante los primeros 10 años de vida y los años posteriores. En los Estados Unidos, antes del advenimiento de la quimiorreducción para tratar la enfermedad hereditaria o bilateral, y antes de la implementación de los exámenes de detección con neuroimágenes, el retinoblastoma trilateral era el factor responsable de más del 50 % de la mortalidad relacionada con el retinoblastoma en los primeros 10 años posteriores al diagnóstico.[30] Para obtener más información sobre NS, consultar la sección Efectos tardíos del tratamiento del retinoblastoma.
Retinoblastoma trilateral
El retinoblastoma trilateral es un síndrome bien reconocido que se presenta en un 5 % a un 15 % de los pacientes con la forma hereditaria del retinoblastoma. Se define por la formación de un tumor neuroblástico en la línea media intracraneal asíncrono que, por lo general, se presenta entre los 20 y 36 meses de vida.[31]
Puesto que el retinoblastoma trilateral tiene un pronóstico adverso, y que parece que la detección temprana y el tratamiento radical mejoran la supervivencia, los exámenes de detección periódicos con neuroimágenes podrían asegurar la detección de la mayoría de casos dentro de los 2 primeros años del diagnóstico inicial de retinoblastoma.[31] Se recomienda hacer de manera rutinaria una IRM encefálica inicial en el momento del diagnóstico porque a veces permite detectar el retinoblastoma trilateral en un estadio subclínico. En una pequeña serie de pacientes, la tasa de supervivencia general a 5 años fue del 67 % en los pacientes con tumores detectados al inicio, en comparación con el 11 % en el grupo de diagnóstico demorado.[32]
Aunque no está claro si el diagnóstico temprano logra mejorar la supervivencia, se ha recomendado la detección con IRM a intervalos de hasta 6 meses durante 5 años en los pacientes en quienes se sospecha una enfermedad hereditaria o que tienen enfermedad unilateral con antecedentes familiares confirmados.[33] Por lo general, se evitan las tomografías computarizadas para la detección de rutina en estos niños debido al riesgo de la exposición a la radiación ionizante.
Una glándula pineal quística, que por lo común se detecta mediante una IRM de vigilancia, se debe diferenciar de una variante quística de pineoblastoma. Se indicó que la incidencia de quistes pineales en niños sin retinoblastoma es del 55,8 %.[34] En un estudio de casos y controles que incluyó a 77 niños con retinoblastoma y 77 controles, la incidencia de quistes pineales fue semejante (61 y 69 %, respectivamente), y el tamaño y volumen de la glándula pineal no varió de manera significativa entre los grupos.[35] Sin embargo, se describió un componente quístico en casi el 57 % de los pacientes con retinoblastoma trilateral confirmado por estudios histológicos.[32] Al parecer el aumento excesivo del tamaño de la glándula pineal es el parámetro más sólido que indica un proceso neoplásico maligno.[35]
Referencias:
Ries LA, Smith MA, Gurney JG, et al., eds.: Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. National Cancer Institute, SEER Program, 1999. NIH Pub.No. 99-4649. Also available online. Last accessed December 22, 2023.
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
Skalet AH, Gombos DS, Gallie BL, et al.: Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists. Ophthalmology 125 (3): 453-458, 2018.
Kamihara J, Bourdeaut F, Foulkes WD, et al.: Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin Cancer Res 23 (13): e98-e106, 2017.
Abramson DH: Re: Skalet et al.: Screening children at risk for retinoblastoma: consensus report from the American Association of Ophthalmic Oncologists and Pathologists (Ophthalmology. 2018;125:453-458). Ophthalmology 125 (9): e63-e64, 2018.
Truong B, Green AL, Friedrich P, et al.: Ethnic, Racial, and Socioeconomic Disparities in Retinoblastoma. JAMA Pediatr 169 (12): 1096-104, 2015.
Khurana A, Eisenhut CA, Wan W, et al.: Comparison of the diagnostic value of MR imaging and ophthalmoscopy for the staging of retinoblastoma. Eur Radiol 23 (5): 1271-80, 2013.
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
Castéra L, Sabbagh A, Dehainault C, et al.: MDM2 as a modifier gene in retinoblastoma. J Natl Cancer Inst 102 (23): 1805-8, 2010.
de Oliveira Reis AH, de Carvalho IN, de Sousa Damasceno PB, et al.: Influence of MDM2 and MDM4 on development and survival in hereditary retinoblastoma. Pediatr Blood Cancer 59 (1): 39-43, 2012.
Shields CL, Dockery P, Ruben M, et al.: Likelihood of Germline Mutation With Solitary Unilateral Retinoblastoma Based on Patient Age at Presentation: Analysis of 482 Consecutive Patients. J Pediatr Ophthalmol Strabismus 58 (6): 355-364, 2021 Nov-Dec.
Andreoli MT, Chau FY, Shapiro MJ, et al.: Epidemiological trends in 1452 cases of retinoblastoma from the Surveillance, Epidemiology, and End Results (SEER) registry. Can J Ophthalmol 52 (6): 592-598, 2017.
Zhang J, Benavente CA, McEvoy J, et al.: A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481 (7381): 329-34, 2012.
Rushlow DE, Mol BM, Kennett JY, et al.: Characterisation of retinoblastomas without RB1 mutations: genomic, gene expression, and clinical studies. Lancet Oncol 14 (4): 327-34, 2013.
McEvoy J, Nagahawatte P, Finkelstein D, et al.: RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 5 (2): 438-50, 2014.
Afshar AR, Pekmezci M, Bloomer MM, et al.: Next-Generation Sequencing of Retinoblastoma Identifies Pathogenic Alterations beyond RB1 Inactivation That Correlate with Aggressive Histopathologic Features. Ophthalmology 127 (6): 804-813, 2020.
Kooi IE, Mol BM, Massink MP, et al.: Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep 6: 25264, 2016.
Francis JH, Richards AL, Mandelker DL, et al.: Molecular Changes in Retinoblastoma beyond RB1: Findings from Next-Generation Sequencing. Cancers (Basel) 13 (1): , 2021.
Ewens KG, Bhatti TR, Moran KA, et al.: Phosphorylation of pRb: mechanism for RB pathway inactivation in MYCN-amplified retinoblastoma. Cancer Med 6 (3): 619-630, 2017.
Richter S, Vandezande K, Chen N, et al.: Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am J Hum Genet 72 (2): 253-69, 2003.
Dommering CJ, Mol BM, Moll AC, et al.: RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J Med Genet 51 (6): 366-74, 2014.
Reddy MA, Butt M, Hinds AM, et al.: Prognostic Information for Known Genetic Carriers of RB1 Pathogenic Variants (Germline and Mosaic). Ophthalmol Retina 5 (4): 381-387, 2021.
Eloy P, Dehainault C, Sefta M, et al.: A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Trp Mutation of RB1. PLoS Genet 12 (2): e1005888, 2016.
Clark R: Retinoblastoma: genetic testing and counseling. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 441-6.
Amitrano S, Marozza A, Somma S, et al.: Next generation sequencing in sporadic retinoblastoma patients reveals somatic mosaicism. Eur J Hum Genet 23 (11): 1523-30, 2015.
Sagi M, Frenkel A, Eilat A, et al.: Genetic screening in patients with Retinoblastoma in Israel. Fam Cancer 14 (3): 471-80, 2015.
Chen Z, Moran K, Richards-Yutz J, et al.: Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat 35 (3): 384-91, 2014.
Nichols KE, Houseknecht MD, Godmilow L, et al.: Sensitive multistep clinical molecular screening of 180 unrelated individuals with retinoblastoma detects 36 novel mutations in the RB1 gene. Hum Mutat 25 (6): 566-74, 2005.
Abramson DH, Mendelsohn ME, Servodidio CA, et al.: Familial retinoblastoma: where and when? Acta Ophthalmol Scand 76 (3): 334-8, 1998.
Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
Sirin S, de Jong MC, Galluzzi P, et al.: MRI-based assessment of the pineal gland in a large population of children aged 0-5 years and comparison with pineoblastoma: part II, the cystic gland. Neuroradiology 58 (7): 713-21, 2016.
Pham TT, Siebert E, Asbach P, et al.: Magnetic resonance imaging based morphologic evaluation of the pineal gland for suspected pineoblastoma in retinoblastoma patients and age-matched controls. J Neurol Sci 359 (1-2): 185-92, 2015.
Características patológicas tumorales del retinoblastoma
El retinoblastoma en humanos surge en células precursoras en maduración de los conos retinianos.[1,2] El aspecto microscópico del retinoblastoma depende del grado de diferenciación. El retinoblastoma indiferenciado se compone de paquetes densos de células pequeñas, redondas, con núcleos hipocromáticos y citoplasma escaso. Se describieron varios grados de diferenciación de los fotorreceptores que se caracterizan por distribuciones diferenciadas de las células tumorales que se describen a continuación:
Las rosetas de Flexner-Wintersteiner son específicas del retinoblastoma. Estas estructuras se componen de un grupo de células cilíndricas cortas, dispuestas alrededor de un lumen central delimitado por una membrana eosinofílica análoga a la membrana externa de la retina normal. El lumen contiene rosetas que se observan en el 70 % de los tumores.
Las rosetas de Homer Wright se componen de anillos irregulares de células tumorales dispuestas alrededor de una maraña de fibrillas sin lumen ni membrana limitante interna. Las rosetas de Homer Wright no se suelen observar en el retinoblastoma y se ven más a menudo en otros tumores neuroblásticos, como el neuroblastoma y el meduloblastoma.
El retinoblastoma se caracteriza por una proliferación celular marcada, como lo demuestra un recuento alto de mitosis, índices de marcación de MIB-1 muy altos, y una inmunorreactividad nuclear fuerte difusa para CRX, un marcador útil para diferenciar el retinoblastoma de otros tumores de células pequeñas redondas malignas.[3,4]
El retinoblastoma cavitario, una variante poco frecuente, tiene cavidades relumbrantes visibles dentro del tumor cuando se observa al examen oftalmoscópico. Los espacios cavitarios tienen un aspecto hueco en la ecografía y son hipofluorescentes en la angiografía. Desde el punto de vista histopatológico, los espacios cavitarios representan áreas de diferenciación de fotorreceptores.[5]
El retinoblastoma cavitario se ha relacionado con una respuesta tumoral visible mínima a la quimioterapia intravenosa e intraarterial, se piensa que esto es un signo de diferenciación tumoral.[6,7] A pesar de la respuesta clínica mitigada, los pacientes con retinoblastoma cavitario tienen desenlaces favorables a largo plazo, con una respuesta tumoral buena y un rescate del globo ocular similar al de los pacientes con retinoblastoma no cavitario.
Es importante que un patólogo con experiencia en patología ocular y retinoblastoma examine las piezas de la enucleación, en especial a fin de determinar las características de riesgo de diseminación extraocular. Para obtener más información, consultar la sección Tratamiento del retinoblastoma intraocular.
Referencias:
Xu XL, Singh HP, Wang L, et al.: Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 514 (7522): 385-8, 2014.
Singh HP, Wang S, Stachelek K, et al.: Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc Natl Acad Sci U S A 115 (40): E9391-E9400, 2018.
Terry J, Calicchio ML, Rodriguez-Galindo C, et al.: Immunohistochemical expression of CRX in extracranial malignant small round cell tumors. Am J Surg Pathol 36 (8): 1165-9, 2012.
Schwimer CJ, Prayson RA: Clinicopathologic study of retinoblastoma including MIB-1, p53, and CD99 immunohistochemistry. Ann Diagn Pathol 5 (3): 148-54, 2001.
Palamar M, Pirondini C, Shields CL, et al.: Cavitary retinoblastoma: ultrasonographic and fluorescein angiographic findings in 3 cases. Arch Ophthalmol 126 (11): 1598-600, 2008.
Mashayekhi A, Shields CL, Eagle RC, et al.: Cavitary changes in retinoblastoma: relationship to chemoresistance. Ophthalmology 112 (6): 1145-50, 2005.
Rojanaporn D, Kaliki S, Bianciotto CG, et al.: Intravenous chemoreduction or intra-arterial chemotherapy for cavitary retinoblastoma: long-term results. Arch Ophthalmol 130 (5): 585-90, 2012.
Sistemas de estadificación y agrupamiento del retinoblastoma
La estadificación de los pacientes con retinoblastoma exige una estrecha coordinación de radiólogos, oncólogos pediatras y oftalmólogos. Se han propuesto varios sistemas de estadificación y agrupamiento del retinoblastoma.[1] La evaluación general de la extensión del retinoblastoma se documenta mediante sistemas de estadificación. La diseminación intraocular, que es importante para el rescate ocular, se documenta mediante sistemas de agrupamiento. Para propósitos de tratamiento, el retinoblastoma se clasifica como enfermedad intraocular y enfermedad extraocular.
Retinoblastoma intraocular
El retinoblastoma intraocular se localiza en el ojo. A veces se limita a la retina, otras veces se disemina hasta comprometer otras estructuras, como la coroides, el cuerpo ciliar, la cámara anterior y la papila óptica. Sin embargo, el retinoblastoma intraocular no se disemina fuera del ojo a los tejidos circundantes ni a otras partes del cuerpo.
Retinoblastoma extraocular
El retinoblastoma extraocular se disemina fuera del ojo. A veces está confinado a los tejidos que rodean el ojo (retinoblastoma orbitario), otras veces se disemina por el sistema nervioso central (SNC), o de manera sistémica hasta la médula ósea o los ganglios linfáticos (retinoblastoma metastásico).
Sistemas de estadificación
Sistema de estadificación del American Joint Committee on Cancer
A lo largo de los años se han propuesto varios sistemas de estadificación. Las nuevas pautas estatales para la notificación del cáncer a los North American Association of Cancer Registries exigen el uso de la estadificación del American Joint Committee on Cancer (AJCC), según la octava edición del manual de estadificación.[2]
Para obtener información sobre las definiciones de la clasificación clínica de tumor primario (T), ganglio linfático regional (N), metástasis a distancia (M), grado histológico y grupos de estadios pronósticos, consultar el Cuadro 3, el Cuadro 5, el Cuadro 7, el Cuadro 8 y el Cuadro 9.
Para obtener información sobre las definiciones de la clasificación patológica, grado histológico y grupos de estadios pronósticos, consultar el Cuadro 4, el Cuadro 6, el Cuadro 7, el Cuadro 8 y el Cuadro 10.
Este sistema de estadificación afecta los casos que se han diagnosticado a partir de 2018. La estadificación del retinoblastoma es la primera en reconocer la función de la predisposición genética al incorporar una categoría H. H1 se refiere a pacientes con retinoblastoma bilateral o trilateral, antecedentes familiares de retinoblastoma o que presentan una variante de RB1 (consultar el Cuadro 2).[2]
Cuadro 2. Definiciones de rasgo hereditario (H)a
Categoría H
Criterios H
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
HX
Datos insuficientes para determinar la presencia de una variante constitucional del genRB1
H0
Alelos deRB1normales en sangre verificados mediante análisis de sensibilidad alta
H1
Retinoblastoma bilateral, retinoblastoma con un tumor neuroectodérmico primitivo intracraneal (es decir, retinoblastoma trilateral), pacientes con historia familiar de retinoblastomao definición molecular de una variante constitucional del genRB1
Cuadro 3. Definiciones clínicas de tumor primario (cT)a
Categoría cT
Criterios cT
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
cTX
Indicios de tumor intraocular desconocidos
cT0
Sin indicios de tumor intraocular
cT1
Uno o más tumores intrarretinales con líquido subretiniano a ≤5 mm de la base de cualquier tumor
cT1a
Tumores ≤3 mm y a más de 1,5 mm de distancia del disco y la fóvea
cT1b
Tumores >3 mm o a menos de 1,5 mm de distancia del disco o de la fóvea
cT2
Uno o más tumores intraoculares con desprendimiento de retina, diseminación vítrea o diseminación subretiniana
cT2a
Líquido subretiniano a >5 mm de la base de cualquier tumor
cT2b
Diseminación vítrea o subretiniana
cT3
Uno o más tumores intraoculares avanzados
cT3a
Atrofia o subatrofia del globo ocular (phthisis bulbi)
cT3b
Invasión tumoral de la coroides, el anillo ciliar (pars plana), el cuerpo ciliar, el cristalino, las zónulas, el iris o la cámara anterior
cT3c
Aumento de la presión intraocular con neovascularización o buftalmía
cT3d
Hipema o hemorragia vítrea masiva
cT3e
Celulitis orbitaria aséptica
cT4
Uno o más tumores extraoculares que afectan a la órbita, incluido el nervio óptico
cT4a
Evidencia radiológica de compromiso del nervio óptico retrobulbar, engrosamiento del nervio óptico o compromiso de los tejidos orbitarios
cT4b
Tumor extraocular clínicamente evidente con proptosis o masa orbitaria
Para una mejor evaluación de la importancia de la diseminación del tumor, en un estudio multicéntrico internacional de registros de personas con ojos con retinoblastoma se investigó si la distribución y las características clínicas de las semillas de diseminación del retinoblastoma observadas en ojos cT2b repercutía sobre el fracaso terapéutico local. De los 624 casos donde se intentó conservar el ojo, 592 tenían datos completos sobre el análisis de conservación del globo ocular. La distribución de las semillas de diseminación fue focal en 143 ojos (24,2 %) y difusa en 449 ojos (75,8 %). En el momento del cuadro clínico inicial, la diseminación difusa se asoció con 2,8 veces más riesgo de fracaso terapéutico en comparación con la presencia de diseminación focal del retinoblastoma. La tasa acumulada de Kaplan-Meier de conservación del globo a 5 años (sin radioterapia externa) fue del 78 % para los ojos con semillas de diseminación focal y del 49 % para los ojos con semillas de diseminación difusas. Esta subclasificación de la diseminación del retinoblastoma no se incluye actualmente en el sistema de estadificación del AJCC.[3][Nivel de evidencia C3]
Cuadro 4. Definiciones patológicas de tumor primario (pT)a
Categoría pT
Criterios pT
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
pTX
Indicios de tumor intraocular desconocidos
pT0
Sin indicios de tumor intraocular
pT1
Uno o más tumores intraoculares sin invasión local, invasión focal de la coroides ni compromiso prelaminar o intralaminar de la papila óptica
pT2
Uno o más tumores intraoculares con invasión local
pT2a
Invasión concomitante focal de la coroides y compromiso prelaminar o intralaminar de la papila óptica
pT2b
Invasión tumoral del estroma del iris, la malla trabecular o el canal de Schelemm
pT3
Uno o más tumores intraoculares con invasión local significativa
pT3a
Invasión masiva de la coroides (>3 mm en su diámetro más grande o focos múltiples de compromiso coroideo focal que afecte en total >3 mm, o cualquier compromiso que abarque el grosor total de la coroides)
pT3b
Invasión retrolaminar de la papila óptica, sin compromiso del extremo transversal del nervio óptico
pT3c
Cualquier compromiso del grosor parcial de la esclerótica dentro de los dos tercios internos
pT3d
Invasión del grosor total de la esclerótica dentro del tercio externo, o invasión dentro o alrededor de los canales emisarios
pT4
Indicios de tumor extraocular: tumor en el extremo transversal del nervio óptico, tumor en los espacios meníngeos alrededor del nervio óptico, invasión del grosor total de la esclerótica con invasión de la epiesclera, el tejido adiposo adyacente, el músculo extraocular, el hueso, la conjuntiva o los parpados
Cuadro 5. Definiciones clínicas de ganglio linfático regional (cN)a
Categoría cN
Criterios cN
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
cNX
Compromiso de ganglios linfáticos regionales no evaluable
cN0
Sin compromiso de ganglios linfáticos
cN1
Evidencia de compromiso de los ganglios linfáticos preauriculares, submandibulares y cervicales
Cuadro 6. Definiciones patológicas de ganglio linfático regional (pN)a
Categoría pN
Criterios pN
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
pNX
Compromiso de ganglios linfáticos regionales no evaluable
pN0
Sin compromiso de ganglios linfáticos
pN1
Compromiso de ganglio linfático regional
Cuadro 7. Definiciones clínicas (c) y patológicas (p)de metástasis a distancia (M)a
Categoría M
Criterios M
SNC = sistema nervioso central.
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
cM0
No hay signos ni síntomas de metástasis a distancia o intracraneal
cM1
Metástasis a distancia sin confirmación microscópica
cM1a
Uno o más tumores que comprometen cualquier sitio a distancia (por ejemplo, médula ósea, hígado) según las pruebas clínicas o radiológicas
cM1b
Tumor que compromete el SNC según imágenes radiológicas (sin incluir al retinoblastoma trilateral)
pM1
Metástasis a distancia con confirmación histopatológica
pM1a
Confirmación histopatológica de tumor en cualquier sitio a distancia (por ejemplo, médula ósea, hígado u otro)
pM1b
Confirmación histopatológica de tumor en el líquido cefalorraquídeo o el parénquima del SNC
Cuadro 8. Grado histológico (G)a
G
Definiciones de G
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
GX
Grado no evaluable
G1
Tumor con áreas de retinoma (organización celular similar a pequeños ramos de flores (fleurettes) o diferenciación neuronal)
G2
Tumor con muchas rosetas (Flexner-Wintersteiner o Homer Wright)
G3
Tumor con rosetas ocasionales (Flexner-Wintersteiner o Homer Wright)
G4
Tumor con células poco diferenciadas sin rosetas o con áreas extensas (más de la mitad del tumor) de anaplasia
Cuadro 9. Grupos de estadios pronósticos del AJCC: Estadio clínico (cTNM)a
cT
N
M
H
Grupo de estadio clínico correspondiente
cM = metástasis a distancia clínica; cN = ganglio linfático regional clínico; cT = tumor primario clínico; H = rasgo hereditario; pM = metástasis a distancia patológica.
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
cT1, cT2, cT3
cN0
cM0
Cualquiera
I
cT4a
cN0
cM0
Cualquiera
II
cT4b
cN0
cM0
Cualquiera
III
Cualquiera
cN1
cM0
Cualquiera
III
Cualquiera
Cualquiera
cM1 o pM1
Cualquiera
IV
Cuadro 10. Grupos de estadios pronósticos del AJCC: Estadio patológico (pTNM)a
pT
N
M
H
Grupo de estadio patológico correspondiente
cM = metástasis a distancia clínica; H = rasgo hereditario; pT = tumor primario patológico; pN = ganglio linfático regional patológico; pM = metástasis a distancia patológica.
a Reproducción autorizada de AJCC: Retinoblastoma. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8th Ed. New York, NY: Springer, 2017, pp. 819–831.
pT1, pT2 o pT3
pN0
cM0
Cualquiera
I
pT4
pN0
cM0
Cualquiera
II
Cualquiera
pN1
cM0
Cualquiera
III
Cualquiera
Cualquiera
cM1 o pM1
Cualquiera
IV
Criterios de tamaño
No hay criterios uniformes sobre el tamaño del retinoblastoma intraocular y su asociación con la presencia de características patológicas de riesgo alto. En una serie de casos internacional, multicéntrica y retrospectiva basada en registros, se analizaron casos de 13 países para evaluar la asociación entre las características patológicas de riesgo alto en el momento del diagnóstico (definidas por los estadios del AJCC pT3 y pT4), las características clínicas de riesgo alto (definidas por los estadios del AJCC cT2 y cT3) y el nuevo sistema de agrupamiento por tamaño propuesto por la AJCC Ophthalmic Oncology Task Force (OOTF), donde se clasificó el tamaño tumoral intraocular en los siguientes 4 grupos:[4][Nivel de evidencia C3]
Grupo de tamaño 1: compromiso de menos de la mitad del globo ocular.
Grupo de tamaño 2: compromiso de más de la mitad, pero menos de dos tercios del globo ocular.
Grupo de tamaño 3: compromiso tumoral de más de dos tercios del globo ocular.
Grupo de tamaño 4: retinoblastoma difuso infiltrante.
De los 942 ojos con retinoblastoma tratados mediante enucleación primaria, 282 (30%) presentaron características patológicas de riesgo alto. Tanto los grupos de estadio clínico (subcategorías cT) como los grupos de tamaño del AJCC se asociaron con características patológicas de riesgo alto.
En comparación con los tumores cT2a (tumores intraoculares con desprendimiento de la retina, semillas de diseminación vítreas o semillas subretinianas con líquido subretiniano >5 mm de cualquier tumor), neovascularización del iris con glaucoma (cT3c) (oportunidad relativa [OR], 2,3), hemorragia intraocular (cT3d) (OR, 2,5), y celulitis orbitaria aséptica (cT3e) (OR, 3,3) fueron factores predictivos de características patológicas de riesgo alto.
En comparación con el grupo de tamaño 1, los grupos de tamaño 3 (OR, 3,3) y 4 (OR, 4,1) fueron los mejores factores de predicción de características patológicas de riesgo alto.
Estos factores clínicos de riesgo pueden, de manera potencial, usarse para predecir la presencia de características patológicas de riesgo alto y facilitar las decisiones sobre el tratamiento.
La misma serie de casos internacional, multicéntrica y retrospectiva basada en registros se utilizó para evaluar el riesgo de muerte por metástasis. El análisis se basó en las características del cuadro clínico inicial (n = 1814 pacientes en estadio clínico cT2 o cT3; n = 1416 pacientes clasificados por tamaño tumoral) y el tratamiento de pacientes con retinoblastoma intraocular avanzado. En este estudio se definió el estadio avanzado como un retinoblastoma de categoría cT2 y cT3 del AJCC y según los grupos de tamaño de AJCC-OOTF. Los tratamientos fueron enucleación primaria, quimioterapia sistémica con enucleación secundaria y quimioterapia sistémica con conservación del ojo.[5][Nivel de evidencia C3]
A mayor subcategoría cT3 mayor riesgo de muerte por metástasis. Se calculó 4,9 veces más riesgo en el grupo cT3c, 14,0 veces más riesgo en el grupo cT3d y 19,6 veces más riesgo en el grupo cT3e en comparación con el riesgo del grupo cT2a.
Otros factores significativos de riesgo de metástasis fueron una mayor edad en el cuadro clínico inicial (mediana de edad en el momento del diagnóstico, 22 meses vs. 16 meses; P < 0,001) y un intento de conservación ocular mediante quimioterapia sistémica.
A mayor grupo de tamaño intraocular mayor riesgo de muerte por metástasis. Se calculó 10,0 veces más riesgo en los pacientes con tumores del grupo de tamaño 3 y 41,1 veces más riesgo en los pacientes con tumores del grupo de tamaño 4 en comparación con el grupo de tamaño 1.
International Retinoblastoma Staging System
El International Retinoblastoma Staging System (IRSS) es un sistema más simple propuesto por un consorcio internacional de oftalmólogos y oncólogos pediatras.[6] El IRSS se emplea de manera más generalizada en el ámbito clínico que el sistema de estadificación del AJCC (consultar el Cuadro 11). En un estudio retrospectivo alemán se encontró que el IRSS permitió predecir la supervivencia de 633 niños con retinoblastoma hereditario, entre ellos, 582 pacientes con enfermedad en estadios IRSS 0 o I.[7]
Cuadro 11. International Retinoblastoma Staging System
Estadio
Descripción
SNC = sistema nervioso central; LCR = líquido cefalorraquídeo.
0
Ojo sin enuclear y sin diseminación de la enfermedad. Para obtener más información, consultar la sección International Classification of Retinoblastoma.
I
Ojo enucleado y resección completa confirmada por examen histológico
II
Ojo enucleado y tumor residual microscópico
III
Diseminación regional
a. Enfermedad orbitaria manifiesta
b. Diseminación a ganglio linfático preauricular o cervical
IV
Enfermedad metastásica
a. Metástasis hematógena (sin compromiso del SNC)
—Lesión única
—Lesiones múltiples
b. Diseminación al SNC (con cualquier otro sitio de enfermedad regional o metastásica regional, o sin estos)
—Lesión prequiasmática
—Masa en el SNC
—Enfermedad leptomeníngea y en el LCR
Sistemas de agrupamiento
Los siguientes sistemas de agrupamiento son importantes para evaluar la diseminación intraocular de la enfermedad y son útiles para predecir el rescate ocular:
International Classification of Retinoblastoma.
American Joint Committee on Cancer (AJCC), octava edición.
Clasificación de los tumores intraoculares de Reese-Ellsworth.
International Classification of Retinoblastoma
El sistema de agrupamiento International Classification of Retinoblastoma se formuló con el objetivo de proporcionar una clasificación más simple y fácil de usar, que pudiera aplicarse a los tratamientos actuales. Este nuevo sistema parte del alcance de la diseminación tumoral dentro de la cavidad vítrea y el espacio subretiniano, en lugar del tamaño y la ubicación del tumor (consultar el Cuadro 12). El uso de este sistema permite predecir mejor el éxito del tratamiento.[8,9,10] Asimismo, el sistema ayuda a predecir el tipo histopatológico de riesgo alto. En un estudio con más de 500 pacientes de retinoblastoma, se observaron hallazgos histopatológicos de enfermedad de riesgo alto en el 17 % de los ojos del grupo D y en el 24 % de los ojos del grupo E. Esto es útil para asesorar a los padres sobre la posible necesidad de tratamiento sistémico posoperatorio.[11]
Cuadro 12. Sistema de agrupamiento de la International Classification of Retinoblastoma
Grupo
Definiciones
Grupo A
Tumores intrarretinianos pequeños fuera de la fovéola y la papila óptica.
Todos los tumores miden 3 mm o menos en su dimensión mayor y se limitan a la retina.
Además, todos los tumores se ubican a más de 3 mm de la fovéola y a más de 1,5 mm de la papila óptica.
Grupo B
Todos los tumores residuales aislados limitados a la retina.
Todos los otros tumores limitados a la retina que no están en el grupo A.
Hay líquido subretiniano relacionado con el tumor a menos de 3 mm del tumor, sin diseminación subretiniana.
Tumor ubicado a menos de 3 mm del nervio óptico o la fóvea.
Grupo C
Enfermedad local aislada con mínima diseminación subretiniana o vítrea.
Uno o más tumores aislados.
Líquido subretiniano presente o pasado, sin diseminación, que compromete hasta un cuarto de la retina.
Es posible que haya diseminación vítrea local fina cerca del tumor aislado.
Diseminación subretiniana local a menos de 3 mm (2 DD) del tumor.
Grupo D
Enfermedad difusa con diseminación vítrea o subretiniana significativas.
Uno o más tumores macizos o difusos.
Líquido subretiniano presente o pasado, sin diseminación, que compromete hasta la totalidad del desprendimiento de retina.
Enfermedad vítrea difusa o maciza que a veces incluye diseminacióngrasosa o masas tumorales avasculares.
Diseminación subretiniana difusa que a veces incluye placas subretinianas o nódulos tumorales.
Grupo E
Presencia de una o más de las siguientes características de pronóstico precario:
Tumor que toca el cristalino.
Tumor anterior a la superficie vítrea anterior con compromiso del cuerpo ciliar o el segmento anterior.
Retinoblastoma infiltrante difuso.
Glaucoma neovascular.
Opacidad de la túnica media por hemorragia.
Necrosis tumoral con celulitis orbitaria aséptica.
Subatrofia del globo ocular.
Clasificación de los tumores intraoculares de Reese-Ellsworth
Reese y Ellsworth formularon un sistema de clasificación para el retinoblastoma intraocular que mostró tener valor predictivo para la conservación de la vista y el control de la enfermedad local en la época en que la cirugía y la radioterapia de haz externo (RHE) eran las opciones principales de tratamiento. Sin embargo, los avances en el tratamiento conservador del retinoblastoma intraocular han hecho que el sistema de clasificación de Reese-Ellsworth sea menos predictivo del rescate ocular y menos útil para guiar el tratamiento.[9] Este sistema de agrupamiento casi no se usa y su utilidad principal es de referencia histórica.
Referencias:
Chantada GL, Sampor C, Bosaleh A, et al.: Comparison of staging systems for extraocular retinoblastoma: analysis of 533 patients. JAMA Ophthalmol 131 (9): 1127-34, 2013.
Mallipatna A, Gallie BL, Chevez-Barrios P, et al.: Retinoblastoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 819-31.
Tomar AS, Finger PT, Gallie B, et al.: Retinoblastoma seeds: impact on American Joint Committee on Cancer clinical staging. Br J Ophthalmol 107 (1): 127-132, 2023.
Tomar AS, Finger PT, Gallie B, et al.: High-risk Pathologic Features Based on Presenting Findings in Advanced Intraocular Retinoblastoma: A Multicenter, International Data-Sharing American Joint Committee on Cancer Study. Ophthalmology 129 (8): 923-932, 2022.
Tomar AS, Finger PT, Gallie B, et al.: Metastatic Death Based on Presenting Features and Treatment for Advanced Intraocular Retinoblastoma: A Multicenter Registry-Based Study. Ophthalmology 129 (8): 933-945, 2022.
Chantada G, Doz F, Antoneli CB, et al.: A proposal for an international retinoblastoma staging system. Pediatr Blood Cancer 47 (6): 801-5, 2006.
Temming P, Arendt M, Viehmann A, et al.: How Eye-Preserving Therapy Affects Long-Term Overall Survival in Heritable Retinoblastoma Survivors. J Clin Oncol 34 (26): 3183-8, 2016.
Murphree L: Staging and grouping of retinoblastoma. In: Singh A, Damato B: Clinical Ophthalmic Oncology. Saunders Elsevier, 2007, pp 422-7.
Shields CL, Mashayekhi A, Au AK, et al.: The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 113 (12): 2276-80, 2006.
Novetsky DE, Abramson DH, Kim JW, et al.: Published international classification of retinoblastoma (ICRB) definitions contain inconsistencies--an analysis of impact. Ophthalmic Genet 30 (1): 40-4, 2009.
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
Aspectos generales de las opciones de tratamiento del retinoblastoma
Para optimizar los desenlaces del tratamiento, es necesario que su planificación esté a cargo de un equipo multidisciplinario de especialistas en cáncer con experiencia en el abordaje de los tumores oculares infantiles, como oncólogos pediatras, oftalmólogos y radioncólogos.[1] Antes de iniciar el tratamiento es muy recomendable hacer una evaluación en centros oncológicos especializados con el fin de aumentar la probabilidad de rescate ocular y conservación de la vista.
Los objetivos del tratamiento son los siguientes:
Erradicar la enfermedad para salvar la vida del paciente.
Conservar la vista tanto como sea posible.
Disminuir el riesgo de secuelas tardías del tratamiento; en particular, neoplasias subsiguientes (NS).
Muchos tratamientos que se consideraban el estándar de atención no se han sometido a estudio con métodos aleatorizados.
El tratamiento del retinoblastoma depende de la carga de enfermedad intraocular y extraocular, la lateralidad de la enfermedad, el estado en la línea germinal del gen RB1 y la posibilidad de conservar la vista. En los pacientes que presentan enfermedad intraocular, en especial, aquellos con compromiso ocular bilateral, es posible que un abordaje conservador de reducción tumoral con quimioterapia intravenosa o intraarterial (quimioterapia en la arteria oftálmica), acompañada de terapia local intensiva, produzca tasas altas de rescate ocular.[2] La radioterapia, uno de los tratamientos más eficaces para el retinoblastoma, se suele reservar para los casos de progresión intraocular o extraocular de la enfermedad.
Se debe considerar una combinación razonable y adaptada al riesgo de las siguientes opciones terapéuticas:
Enucleación.
Tratamiento local (crioterapia, terapia láser y braquiterapia).
Quimioterapia sistémica.
Quimioterapia intraarterial (infusión de quimioterapia en la arteria oftálmica).
Quimioterapia intravítrea.
Quimioterapia intracameral.
Radioterapia (radioterapia de haz externo [RHE], braquiterapia).
Las opciones de tratamiento del retinoblastoma intraocular, extraocular y recidivante se describen en el Cuadro 13.
Cuadro 13. Opciones de tratamiento del retinoblastoma
Grupo de tratamiento
Opciones de tratamiento
RHE = radioterapia de haz externo; SNC = sistema nervioso central.
Retinoblastoma intraocular:
Retinoblastoma unilateral
Enucleación de tumores intraoculares grandes, con quimioterapia adyuvante o sin esta
Abordajes conservadores de rescate ocular cuando es posible salvar el ojo y la vista:
—Quimiorreducción y quimioterapia sistémica o quimioterapia intraarterial con quimioterapia intravítrea o sin esta
—Tratamientos locales (crioterapia, termoterapia y radioterapia con placas)
Retinoblastoma bilateral
Enucleación para tumores intraoculares grandes, seguida de quimioterapia adaptada al riesgo y con base en las características patológicas cuando no es posible salvar el ojo ni la vista
Abordajes conservadores de rescate ocular cuando es posible salvar el ojo y la vista:
—Quimiorreducción y quimioterapia sistémica o quimioterapia intraarterial con quimioterapia intravítrea o sin esta
—Tratamientos locales (crioterapia, termoterapia y radioterapia con placas)
—RHE
Retinoblastoma extraocular:
Retinoblastoma orbitario y locorregional
Quimioterapia
Radioterapia
Enucleación(para diseminación extraocular)
Enfermedad en el SNC
Quimioterapia sistémica y terapia dirigida al SNC con radioterapia
Quimioterapia sistémica seguida de quimioterapia mielosupresora con rescate de células madre y radioterapia o sin esta
Retinoblastoma trilateral sincrónico
Quimioterapia sistémica seguida de cirugía y quimioterapia mielosupresora con rescate de células madre
Quimioterapia sistémica seguida de cirugía y radioterapia
Retinoblastoma extracraneal metastásico
Quimioterapia sistémica seguida de quimioterapia mielosupresora con rescate de células madre y radioterapia
Retinoblastoma intraocular progresivo o recidivante
Enucleación
Radioterapia (RHE o radioterapia con placas)
Tratamientos locales (crioterapia o termoterapia)
Quimioterapia de rescate (sistémica o intraarterial)
Quimioterapia intravítrea, en especial para la diseminación vítrea resistente al tratamiento o recidivante
Retinoblastoma extraocular progresivo o recidivante
Quimioterapia sistémica y radioterapia para la enfermedad orbitaria
Quimioterapia sistémica seguida de quimioterapia mielosupresora con rescate de células madre y radioterapia para la enfermedad extraorbitaria
Enucleación
Se indica la resección inicial del ojo para tumores grandes que llenan el cuerpo vítreo con poca o ninguna probabilidad de restaurar la visión, y en casos de diseminación a la cámara anterior o en presencia de glaucoma neovascular. Se debe vigilar atentamente a los pacientes para detectar una recidiva orbitaria de la enfermedad, sobre todo durante los 2 primeros años después de la enucleación.[3][Nivel de evidencia C1]
La enucleación también se usa como tratamiento de rescate en casos de progresión o recidiva de la enfermedad en pacientes tratados con abordaje de rescate ocular. La pieza de patología se debe examinar minuciosamente para identificar a los pacientes en riesgo alto de diseminación extraocular que podrían necesitar quimioterapia adyuvante.[4][Nivel de evidencia C1 y C2]
La enucleación en pacientes menores de 3 años no permite el crecimiento adecuado de la órbita durante el desarrollo que sigue, lo que produce asimetría en el tamaño final de la órbita.[5]
Tratamiento local (crioterapia, terapia láser y braquiterapia)
Los pacientes sometidos a tratamientos de rescate ocular siempre necesitan terapia local intensiva. Un oftalmólogo aplica el tratamiento local directamente en el tumor.
Crioterapia. Este tratamiento se basa en la aplicación de una criosonda en la esclerótica en la vecindad inmediata al tumor retiniano. Se usa como tratamiento primario o con quimioterapia para tumores menores de 4 diámetros del disco óptico (DD) ubicados en la parte anterior de la retina.
Terapia láser. Esta opción a veces se usa como tratamiento primario de tumores pequeños, o se combina con quimioterapia para tumores más grandes. La fotocoagulación tradicional (láser de argón), en la que se aplicaba el rayo láser alrededor del tumor dirigido a la vasculatura tumoral se dejó de lado y se dio paso a la termoterapia (láser de diodo). La termoterapia se administra directamente en la superficie del tumor mediante rayos de longitud de onda en el espectro infrarrojo.[6,7]
Braquiterapia (radioterapia con placas). Se utiliza para tumores grandes que no son susceptibles a la crioterapia o la terapia con láser, este tipo de terapia proporciona un mecanismo eficaz para el control local. Para obtener más información, consultar la sección Radioterapia.
Quimioterapia sistémica
La quimioterapia sistémica cumple una función en los siguientes casos:
Entorno adyuvante para pacientes con características patológicas de riesgo alto. Se han utilizado diferentes regímenes de tratamiento para los pacientes con características patológicas de riesgo alto en la pieza de enucleación. La mayoría de los regímenes terapéuticos incluyen una combinación de tres fármacos con vincristina, etopósido y carboplatino, solos o en alternancia con ciclofosfamida y una antraciclina.[8,9,10,11]; [4][Nivel de evidencia C1 y C2]
Tratamiento de pacientes con enfermedad extraocular y metastásica. Los pacientes con enfermedad extraocular se benefician de un tratamiento más intensivo. Aunque no se ha establecido el tratamiento estándar, se informó de reacciones favorables a los regímenes a base de cisplatino con consolidación de dosis altas de quimioterapia y rescate autógeno de células madre hematopoyéticas para los pacientes que tienen enfermedad extraorbitaria.[12,13,14,15]
Tratamiento quimiorreductor combinado con terapia local intensiva para pacientes sometidos a tratamiento de rescate ocular. Durante los últimos 20 años, el estándar de atención ha sido la quimioterapia sistémica para reducir el volumen tumoral (quimiorreducción), facilitar la administración de tratamientos locales y evitar los efectos a largo plazo de la radioterapia.[16] La tasa de éxito del rescate ocular difiere entre las instituciones, pero, en general, se han obtenido desenlaces oculares favorables en tumores aislados sin diseminación vítrea.
En un análisis de una cohorte numerosa de 994 ojos en 554 pacientes que se trataron con quimioterapia intravenosa y de los que se contaba con datos de desenlaces a largo plazo, los investigadores observaron que el control del tumor dependía en gran medida de la designación del grupo por ojo de la International Classification of Retinoblastoma. La quimioterapia intravenosa de primera línea que consiste en 6 ciclos de vincristina, etopósido y carboplatino con quimioterapia intraarterial adicional o radioterapia con placas produjo el control tumoral de los grupos A (96 %), B (91 %), C (91 %), D (71 %) y E (32 %) antes del fin del segundo año. Con el tratamiento mencionado, se pudo evitar la enucleación o la radioterapia de haz externo, y el efecto de control del tumor duró hasta 20 años.[17][Nivel de evidencia C1]
Cuando se utiliza este abordaje, el mejor predictor del rescate ocular es el grupo ocular según la definición de la International Classification of Retinoblastoma; las tasas de rescate oscilan entre el 60 % y el 100 %.[16]
En el contexto de enfermedad con actividad intraocular persistente, se debe ser precavido al usar un abordaje de prolongación de la quimioterapia en lugar de enucleación, porque este abordaje se relacionó con aumento del riesgo de enfermedad metastásica.[18]; [19][Nivel de evidencia C2]
Quimioterapia intraarterial (infusión de quimioterapia en la arteria oftálmica)
La administración directa de quimioterapia en el ojo mediante canulación de la arteria oftálmica es un método factible y eficaz para el rescate ocular cuando se realiza en centros con volumen alto de pacientes que cuentan con servicios especializados de radiólogos intervencionistas expertos en esta área y anestesiología pediátrica. El Children's Oncology Group dirigió un estudio multiinstitucional (ARET12P1 [NCT02097134]) para evaluar la factibilidad de administrar terapia intraarterial en pacientes con diagnóstico nuevo de retinoblastoma del grupo D. El estudio no logró las metas de factibilidad lo que resalta la importancia de derivar a los pacientes a instituciones que atienden números altos de pacientes y tienen experiencia en este procedimiento.[20] Es posible consolidar aún más las respuestas a la quimioterapia con este abordaje al seguir las medidas de control local descritas antes.
El melfalán es el fármaco que se utiliza con mayor frecuencia y es el más eficaz para la quimioterapia intraarterial. A menudo, se combina con topotecán o carboplatino cuando las respuestas fueron subóptimas o hay enfermedad intraocular muy avanzada.[21,22]
El desenlace tras la quimioterapia intraarterial se relaciona con el grado de compromiso intraocular de la siguiente manera:
Los pacientes con enfermedad intraocular temprana (ojos en los grupos B y C) tienen desenlaces satisfactorios con tasas de rescate ocular superiores al 85 % y se les puede tratar con monoterapia.[22]
Los pacientes con ojos clasificados en el grupo D tienen un pronóstico más precario y tasas de rescate ocular inferiores al 60 %.[22] Sin embargo, en centros especializados se han notificado tasas de rescate ocular superiores al 80 %.[21,23] En los pacientes con enfermedad intraocular muy avanzada, una alternativa de tratamiento es emplear quimioterapia sistémica seguida de consolidación con melfalán intraarterial.[24]
Las tasas de rescate ocular cuando se administra quimioterapia intraarterial como tratamiento de rescate para los pacientes con enfermedad recidivante o progresiva son, de modo sistemático más bajas, con tasas de supervivencia general del 50 % al 75 %.[21,22,23] Los mejores resultados se obtienen mediante un régimen más intensivo de tres fármacos con melfalán, topotecán y carboplatino.[25]
La función de la quimioterapia intraarterial para el rescate ocular se ha aclarado más en un ensayo clínico multicéntrico. En este ensayo se comparó la quimioterapia intraarterial con la quimioterapia sistémica en niños con retinoblastoma unilateral avanzado (grupos D o E). Los pacientes se asignaron al azar a recibir 4 ciclos de quimioterapia combinada con melfalán intraarterial (2 ciclos de carboplatino y 2 ciclos de topotecán) o 6 ciclos de quimioterapia sistémica con vincristina, carboplatino y etopósido. El control local se notificó según la práctica estándar. Las tasas de rescate ocular sin progresión a 2 años fueron del 53 % en los pacientes del grupo de quimioterapia intraarterial y del 27 % en los pacientes del grupo de quimioterapia intravenosa. Las tasas de rescate ocular fueron del 71 % en los pacientes que recibieron quimioterapia intraarterial y del 51 % en los pacientes que recibieron quimioterapia intravenosa.[26]
Los pacientes con enfermedad bilateral se pueden someter a una administración intraarterial en tándem.[27] En tal caso, los pacientes tienen un riesgo más alto de toxicidad sistémica causada por el melfalán,[28] y es posible emplear carboplatino en monoterapia para tratar el ojo con enfermedad menos avanzada durante el procedimiento en tándem.[29] En los neonatos y lactantes muy pequeños en quienes la canulación de la arteria oftálmica no es viable, se observó que es muy eficaz el tratamiento puente con carboplatino sistémico en monoterapia hasta que el lactante cumpla 3 meses o pese 6 kg, seguido de consolidación con quimioterapia intraarterial. En un estudio, la tasa de supervivencia ocular sin radiación a 1 año fue del 95 %.[30]
En un estudio de 39 lactantes menores de 3 meses con retinoblastoma intraocular en estadio avanzado (grupos de ojos D y E), los pacientes recibieron quimioterapia intraarterial como tratamiento primario (29 ojos) o tratamiento secundario (13 ojos tratados antes con quimioterapia intravenosa) mediante un procedimiento de microcateterismo. Se utilizó la arteria meníngea media cuando no se pudo cateterizar la arteria oftálmica.[31]
El rescate del globo ocular fue del 96 % para 23 ojos del grupo D y del 33 % para 6 ojos del grupo E; sin embargo, la tasa supervivencia ocular general fue del 81 % debido a un aumento de la incidencia de subatrofia del globo ocular.
La incidencia de ceguera total fue alta.
Como el seguimiento fue limitado y osciló entre 6 meses y 6 años, no se conocen los efectos tardíos.
Según parece, la adición de quimioterapia intravítrea a la quimioterapia intraarterial mejora de manera considerable la eficacia general del tratamiento en los ojos con diseminación vítrea; en especial, cuando hay diseminación vítrea difusa.[21,32,33] Para obtener más información, consultar la sección Quimioterapia intravítrea.
En los pacientes que tienen un desprendimiento de retina completo, la quimiocirugía de la arteria oftálmica demostró promover la reconexión de la retina.[34]
Las complicaciones relacionadas con la quimioterapia intraarterial son las siguientes:[22,26,35]
Efectos vasculares e isquémicos, incluso la estenosis (18 %) y la oclusión de la arteria oftálmica (hasta el 9 %).
Atrofia óptica (3,4 %).
Subatrofia del globo ocular (2,7 %).
Las complicaciones vasculares graves relacionadas con el procedimiento son muy infrecuentes. Los grupos de mayor experiencia no han notificado accidentes cerebrovasculares ni complicaciones neurológicas agudas de importancia.[21,22,37] Sin embargo, se documentó estenosis de la arteria oftálmica y oclusión de la arteria central de la retina;[35,37]. El riesgo de trombosis aumenta de manera significativa en niños con trombofilias.[38] En una serie grande de 196 pacientes tratados con 682 infusiones de quimioterapia intraarterial, se notificaron complicaciones vasculares oftálmicas en el 17 % de los ojos tratados.[35]
No se ha evaluado por completo el efecto de los cambios vasculares intraoculares en la visión debido a la corta edad de las primeras cohortes de pacientes tratados. La mayoría de los pacientes no presentan cambios electroretinográficos importantes,[39] y se notificó conservación de la visión central.[40] Es posible que una proporción de pacientes con electrorretinogramas (ERG) anormales, con desprendimiento de retina o sin este, mejoren su ERG en los años que siguen a la quimioterapia intraarterial.[41] No obstante, la quimioterapia intraarterial a veces empeora el funcionamiento de la retina en los pacientes que recibieron tratamientos oculares muy intensivos.[25]
Otro riesgo relacionado con la quimioterapia intraarterial es la exposición a la radiación ionizante durante la fluoroscopia. Se notificó una media de la dosis total de radiación de 42,3 mGy en centros con bastante experiencia.[42] Después de múltiples procedimientos, las dosis acumuladas alcanzan 0,1 a 0,2 Gy, lo que podría causar cataratas y un efecto carcinogénico en esta población susceptible.[43] No ha habido aumento de la incidencia de segundas neoplasias malignas.[44,45] Sin embargo, se necesita un seguimiento más prolongado para definir mejor los riesgos relacionados con este procedimiento.
El riesgo de progresión metastásica con la administración ocular directa parece ser muy bajo.[2] No obstante, se notificaron hasta 20 casos de pacientes tratados con quimioterapia intraarterial que luego presentaron metástasis.[22]
Quimioterapia intravítrea
En estudios se indicó que la inyección intravítrea directa de melfalán o topotecán quizá sea eficaz para neutralizar la diseminación activa en el vítreo.[46,47]; [48,49][Nivel de evidencia C2] En un estudio retrospectivo de 264 ojos (250 niños) tratados con melfalán intravítreo para el control de la diseminación vítrea durante un período de 20 años, se informó de una tasa de remisión completa del 68 %. La incidencia de diseminación extraocular fue baja y se presentó en los niños que tenían características de riesgo alto.[50][Nivel de evidencia C2]
Debido a preocupaciones iniciales sobre el potencial de diseminación tumoral se ha limitado el uso de la quimioterapia intravítrea. Sin embargo, en revisiones adicionales se calculó que la proporción de pacientes que presentan diseminación tumoral extraocular, posiblemente como consecuencia de la inyección en el cuerpo vítreo, es insignificante.[51,52] Si bien este procedimiento es inocuo y se tolera bien, en estudios recientes se demostró una correlación directa entre el número de inyecciones y la disminución en el funcionamiento de la retina, según se midió en el ERG.[52]; [53][Nivel de evidencia C3]
En datos preliminares se apoya el uso de la quimioterapia intraarterial con quimioterapia intravítrea (según indicación por diseminación vítrea) porque es posible que mejore el rescate del globo ocular en ojos con retinoblastoma avanzado en comparación con niños tratados con quimioterapia intraarterial sola en años anteriores.[52]; [32][Nivel de evidencia C2] En comparación con los niños que se trataron en una época anterior, los niños que se trataron en esta última época recibieron una combinación de quimioterapia intraarterial e intravítrea que se relacionó con un tiempo más corto hasta la regresión, menos recidivas, menos enucleaciones y ausencia de aumento de toxicidad, sin diferencias en el deterioro del funcionamiento de la retina evaluado por ERG.[33][Nivel de evidencia C3]
A medida que aumentó la experiencia con el uso de la quimioterapia intravítrea, en los estudios se demostró su eficacia para el control de la diseminación subretiniana y los tumores recidivantes de retina, lo que indica una posible función más allá del control de la diseminación vítrea, como terapia auxiliar para el tratamiento de conservación del globo ocular en el retinoblastoma.[54]
Quimioterapia intracameral
En un estudio retrospectivo de una sola institución se notificó acerca de un tratamiento alternativo para la diseminación a la cámara anterior con inyección de melfalán en el humor acuoso. Se logró la recuperación ocular en 6 de 11 ojos (mediana, 4 inyecciones), con una media de seguimiento de 17 meses.[55]
Radioterapia
El retinoblastoma es una neoplasia maligna muy sensible a la radiación.
RHE. Las dosis de RHE que oscilan entre 35 y 46 Gy por lo general producen remisiones a largo plazo. Es importante contar con una pericia especial en la administración de radioterapia pediátrica, debido a la necesidad de sedar a los niños pequeños y a los detalles intrincados de la planificación del campo. La radioterapia se usa en casos de progresión después de abordajes conservadores en pacientes con diseminación extraocular y como parte del tratamiento de los pacientes con enfermedad metastásica.
Los métodos más recientes de administración de la RHE se aplican para reducir los efectos adversos a largo plazo. Estos son la radioterapia de intensidad modulada y la radioterapia con haz de protones (radioterapia con partículas cargadas).[56,57,58,59] Los datos preliminares indican que la radioterapia de protones se vincula con un riesgo más bajo de neoplasias malignas causadas por la radiación en los sobrevivientes de retinoblastoma hereditario.
En un estudio no aleatorizado en el que se compararon 2 cohortes contemporáneas de pacientes de retinoblastoma hereditario tratados con radioterapia con fotones o radioterapia con protones, la incidencia acumulada de 10 años de NS inducidas por radiación fue significativamente diferente entre los 2 grupos (0 % para la radiación de protones vs. 14 % para la radiación de fotones, P = 0,015).[60]
La RHE administrada a lactantes impide el crecimiento de los huesos orbitarios y produce deformidades cosméticas. La RHE también aumenta el riesgo de NS en niños con retinoblastoma hereditario.
Braquiterapia (radioterapia con placas). Una de las indicaciones de la radioterapia con placas son los tumores solitarios con un diámetro que oscila entre 6 y 15 mm, grosor de 10 mm o menos y ubicación del tumor a más de 3 mm de la papila óptica o fovéola. El radioisótopo que se usa con mayor frecuencia es el yodo I 125, aunque otros como el iridio Ir 192 y el rutenio Ru 106 también son eficaces. En combinación con el uso adecuado de quimioterapia y otras formas de consolidación focal, la braquiterapia quizás sea muy eficaz para el tratamiento de tumores localizados en la retina que no son susceptibles a otros métodos de terapia local.[61,62,63]
Referencias:
Chintagumpala M, Chevez-Barrios P, Paysse EA, et al.: Retinoblastoma: review of current management. Oncologist 12 (10): 1237-46, 2007.
Abramson DH, Fabius AW, Issa R, et al.: Advanced Unilateral Retinoblastoma: The Impact of Ophthalmic Artery Chemosurgery on Enucleation Rate and Patient Survival at MSKCC. PLoS One 10 (12): e0145436, 2015.
Kim JW, Kathpalia V, Dunkel IJ, et al.: Orbital recurrence of retinoblastoma following enucleation. Br J Ophthalmol 93 (4): 463-7, 2009.
Chévez-Barrios P, Eagle RC, Krailo M, et al.: Study of Unilateral Retinoblastoma With and Without Histopathologic High-Risk Features and the Role of Adjuvant Chemotherapy: A Children's Oncology Group Study. J Clin Oncol 37 (31): 2883-2891, 2019.
Oatts JT, Robbins JA, de Alba Campomanes AG: The effect of enucleation on orbital growth in patients with retinoblastoma. J AAPOS 21 (4): 309-312, 2017.
Shields CL, Santos MC, Diniz W, et al.: Thermotherapy for retinoblastoma. Arch Ophthalmol 117 (7): 885-93, 1999.
Francis JH, Abramson DH, Brodie SE, et al.: Indocyanine green enhanced transpupillary thermotherapy in combination with ophthalmic artery chemosurgery for retinoblastoma. Br J Ophthalmol 97 (2): 164-8, 2013.
Chantada GL, Fandiño AC, Guitter MR, et al.: Results of a prospective study for the treatment of unilateral retinoblastoma. Pediatr Blood Cancer 55 (1): 60-6, 2010.
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
Sullivan EM, Wilson MW, Billups CA, et al.: Pathologic risk-based adjuvant chemotherapy for unilateral retinoblastoma following enucleation. J Pediatr Hematol Oncol 36 (6): e335-40, 2014.
Dunkel IJ, Khakoo Y, Kernan NA, et al.: Intensive multimodality therapy for patients with stage 4a metastatic retinoblastoma. Pediatr Blood Cancer 55 (1): 55-9, 2010.
Matsubara H, Makimoto A, Higa T, et al.: A multidisciplinary treatment strategy that includes high-dose chemotherapy for metastatic retinoblastoma without CNS involvement. Bone Marrow Transplant 35 (8): 763-6, 2005.
Rodriguez-Galindo C, Wilson MW, Haik BG, et al.: Treatment of metastatic retinoblastoma. Ophthalmology 110 (6): 1237-40, 2003.
Kremens B, Wieland R, Reinhard H, et al.: High-dose chemotherapy with autologous stem cell rescue in children with retinoblastoma. Bone Marrow Transplant 31 (4): 281-4, 2003.
Rodriguez-Galindo C, Orbach DB, VanderVeen D: Retinoblastoma. Pediatr Clin North Am 62 (1): 201-23, 2015.
Shields CL, Bas Z, Tadepalli S, et al.: Long-term (20-year) real-world outcomes of intravenous chemotherapy (chemoreduction) for retinoblastoma in 964 eyes of 554 patients at a single centre. Br J Ophthalmol 104 (11): 1548-1555, 2020.
Zhao J, Dimaras H, Massey C, et al.: Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J Clin Oncol 29 (7): 845-51, 2011.
Berry JL, Kogachi K, Aziz HA, et al.: Risk of metastasis and orbital recurrence in advanced retinoblastoma eyes treated with systemic chemoreduction versus primary enucleation. Pediatr Blood Cancer 64 (4): , 2017.
Chintagumpala M, Piao J, Gombos D, et al.: A multi-institutional feasibility study of intra-arterial chemotherapy in children with retinoblastoma. A Children's Oncology Group study (COG ARET12P1). Pediatr Blood Cancer 71 (1): e30718, 2024.
Francis JH, Levin AM, Zabor EC, et al.: Ten-year experience with ophthalmic artery chemosurgery: Ocular and recurrence-free survival. PLoS One 13 (5): e0197081, 2018.
Yousef YA, Soliman SE, Astudillo PPP, et al.: Intra-arterial Chemotherapy for Retinoblastoma: A Systematic Review. JAMA Ophthalmol 134 (5): 584-591, 2016.
Abramson DH, Daniels AB, Marr BP, et al.: Intra-Arterial Chemotherapy (Ophthalmic Artery Chemosurgery) for Group D Retinoblastoma. PLoS One 11 (1): e0146582, 2016.
Shields CL, Kaliki S, Al-Dahmash S, et al.: Management of advanced retinoblastoma with intravenous chemotherapy then intra-arterial chemotherapy as alternative to enucleation. Retina 33 (10): 2103-9, 2013 Nov-Dec.
Marr BP, Brodie SE, Dunkel IJ, et al.: Three-drug intra-arterial chemotherapy using simultaneous carboplatin, topotecan and melphalan for intraocular retinoblastoma: preliminary results. Br J Ophthalmol 96 (10): 1300-3, 2012.
Wen X, Fan J, Jin M, et al.: Intravenous versus super-selected intra-arterial chemotherapy in children with advanced unilateral retinoblastoma: an open-label, multicentre, randomised trial. Lancet Child Adolesc Health 7 (9): 613-620, 2023.
Abramson DH, Marr BP, Francis JH, et al.: Simultaneous Bilateral Ophthalmic Artery Chemosurgery for Bilateral Retinoblastoma (Tandem Therapy). PLoS One 11 (6): e0156806, 2016.
Schaiquevich P, Buitrago E, Taich P, et al.: Pharmacokinetic analysis of melphalan after superselective ophthalmic artery infusion in preclinical models and retinoblastoma patients. Invest Ophthalmol Vis Sci 53 (7): 4205-12, 2012.
Francis JH, Gobin YP, Brodie SE, et al.: Experience of intra-arterial chemosurgery with single agent carboplatin for retinoblastoma. Br J Ophthalmol 96 (9): 1270-1, 2012.
Gobin YP, Dunkel IJ, Marr BP, et al.: Combined, sequential intravenous and intra-arterial chemotherapy (bridge chemotherapy) for young infants with retinoblastoma. PLoS One 7 (9): e44322, 2012.
Chen Q, Zhang B, Dong Y, et al.: Intra-arterial chemotherapy as primary or secondary treatment for infants diagnosed with advanced retinoblastoma before 3 months of age. BMC Cancer 19 (1): 693, 2019.
Shields CL, Alset AE, Say EA, et al.: Retinoblastoma Control With Primary Intra-arterial Chemotherapy: Outcomes Before and During the Intravitreal Chemotherapy Era. J Pediatr Ophthalmol Strabismus 53 (5): 275-84, 2016.
Francis JH, Iyer S, Gobin YP, et al.: Retinoblastoma Vitreous Seed Clouds (Class 3): A Comparison of Treatment with Ophthalmic Artery Chemosurgery with or without Intravitreous and Periocular Chemotherapy. Ophthalmology 124 (10): 1548-1555, 2017.
Rowlands MA, Mondesire-Crump I, Levin A, et al.: Total retinal detachments due to retinoblastoma: Outcomes following intra-arterial chemotherapy/ophthalmic artery chemosurgery. PLoS One 13 (4): e0195395, 2018.
Ancona-Lezama D, Dalvin LA, Lucio-Alvarez JA, et al.: OPHTHALMIC VASCULAR EVENTS AFTER INTRA-ARTERIAL CHEMOTHERAPY FOR RETINOBLASTOMA: Real-World Comparison Between Primary and Secondary Treatments. Retina 39 (12): 2264-2272, 2019.
Shields CL, Say EAT, Pefkianaki M, et al.: RHEGMATOGENOUS RETINAL DETACHMENT AFTER INTRAARTERIAL CHEMOTHERAPY FOR RETINOBLASTOMA: The 2016 Founders Award Lecture. Retina 37 (8): 1441-1450, 2017.
Francis JH, Gobin YP, Nagiel A, et al.: Thrombophilia in patients with retinoblastoma receiving ophthalmic artery chemosurgery. Arch Ophthalmol 130 (12): 1605-8, 2012.
Abramson DH: Chemosurgery for retinoblastoma: what we know after 5 years. Arch Ophthalmol 129 (11): 1492-4, 2011.
Brodie SE, Munier FL, Francis JH, et al.: Persistence of retinal function after intravitreal melphalan injection for retinoblastoma. Doc Ophthalmol 126 (1): 79-84, 2013.
Abdelhakim AH, Francis JH, Marr BP, et al.: Retinal reattachment and ERG recovery after ophthalmic artery chemosurgery for advanced retinoblastoma in eyes with minimal baseline retinal function. Br J Ophthalmol 101 (5): 623-628, 2017.
Boddu SR, Abramson DH, Marr BP, et al.: Selective ophthalmic artery chemosurgery (SOAC) for retinoblastoma: fluoroscopic time and radiation dose parameters. A baseline study. J Neurointerv Surg 9 (11): 1107-1112, 2017.
Vijayakrishnan R, Shields CL, Ramasubramanian A, et al.: Irradiation toxic effects during intra-arterial chemotherapy for retinoblastoma: should we be concerned? Arch Ophthalmol 128 (11): 1427-31, 2010.
Suzuki S, Yamane T, Mohri M, et al.: Selective ophthalmic arterial injection therapy for intraocular retinoblastoma: the long-term prognosis. Ophthalmology 118 (10): 2081-7, 2011.
Habib LA, Francis JH, Fabius AW, et al.: Second primary malignancies in retinoblastoma patients treated with intra-arterial chemotherapy: the first 10 years. Br J Ophthalmol 102 (2): 272-275, 2018.
Francis JH, Abramson DH, Gaillard MC, et al.: The classification of vitreous seeds in retinoblastoma and response to intravitreal melphalan. Ophthalmology 122 (6): 1173-9, 2015.
Shields CL, Manjandavida FP, Arepalli S, et al.: Intravitreal melphalan for persistent or recurrent retinoblastoma vitreous seeds: preliminary results. JAMA Ophthalmol 132 (3): 319-25, 2014.
Ghassemi F, Shields CL: Intravitreal melphalan for refractory or recurrent vitreous seeding from retinoblastoma. Arch Ophthalmol 130 (10): 1268-71, 2012.
Munier FL, Gaillard MC, Balmer A, et al.: Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol 96 (8): 1078-83, 2012.
Suzuki S, Aihara Y, Fujiwara M, et al.: Intravitreal injection of melphalan for intraocular retinoblastoma. Jpn J Ophthalmol 59 (3): 164-72, 2015.
Smith SJ, Smith BD: Evaluating the risk of extraocular tumour spread following intravitreal injection therapy for retinoblastoma: a systematic review. Br J Ophthalmol 97 (10): 1231-6, 2013.
Francis JH, Brodie SE, Marr B, et al.: Efficacy and Toxicity of Intravitreous Chemotherapy for Retinoblastoma: Four-Year Experience. Ophthalmology 124 (4): 488-495, 2017.
Smith SJ, Smith BD, Mohney BG: Ocular side effects following intravitreal injection therapy for retinoblastoma: a systematic review. Br J Ophthalmol 98 (3): 292-7, 2014.
Abramson DH, Ji X, Francis JH, et al.: Intravitreal chemotherapy in retinoblastoma: expanded use beyond intravitreal seeds. Br J Ophthalmol 103 (4): 488-493, 2019.
Munier FL, Moulin A, Gaillard MC, et al.: Intracameral Chemotherapy for Globe Salvage in Retinoblastoma with Secondary Anterior Chamber Invasion. Ophthalmology 125 (4): 615-617, 2018.
Krasin MJ, Crawford BT, Zhu Y, et al.: Intensity-modulated radiation therapy for children with intraocular retinoblastoma: potential sparing of the bony orbit. Clin Oncol (R Coll Radiol) 16 (3): 215-22, 2004.
Reisner ML, Viégas CM, Grazziotin RZ, et al.: Retinoblastoma--comparative analysis of external radiotherapy techniques, including an IMRT technique. Int J Radiat Oncol Biol Phys 67 (3): 933-41, 2007.
Lee CT, Bilton SD, Famiglietti RM, et al.: Treatment planning with protons for pediatric retinoblastoma, medulloblastoma, and pelvic sarcoma: how do protons compare with other conformal techniques? Int J Radiat Oncol Biol Phys 63 (2): 362-72, 2005.
Mouw KW, Sethi RV, Yeap BY, et al.: Proton radiation therapy for the treatment of retinoblastoma. Int J Radiat Oncol Biol Phys 90 (4): 863-9, 2014.
Sethi RV, Shih HA, Yeap BY, et al.: Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer 120 (1): 126-33, 2014.
Shields CL, Shields JA, Cater J, et al.: Plaque radiotherapy for retinoblastoma: long-term tumor control and treatment complications in 208 tumors. Ophthalmology 108 (11): 2116-21, 2001.
Merchant TE, Gould CJ, Wilson MW, et al.: Episcleral plaque brachytherapy for retinoblastoma. Pediatr Blood Cancer 43 (2): 134-9, 2004.
Shields CL, Mashayekhi A, Sun H, et al.: Iodine 125 plaque radiotherapy as salvage treatment for retinoblastoma recurrence after chemoreduction in 84 tumors. Ophthalmology 113 (11): 2087-92, 2006.
Consideraciones especiales para el tratamiento de niños con cáncer
El cáncer en niños y adolescentes es raro, aunque desde 1975 se ha observado un aumento gradual de la incidencia general.[1] Los niños y adolescentes con cáncer se deben derivar a centros médicos que cuenten con equipos multidisciplinarios de especialistas en oncología con experiencia en el tratamiento de los cánceres que se presentan en la niñez y la adolescencia. Este equipo multidisciplinario incorpora la pericia de los siguientes profesionales de atención de la salud y otros para asegurar que los niños reciban el tratamiento, los cuidados médicos de apoyo y la rehabilitación que les permitan lograr una supervivencia y calidad de vida óptimas:
La American Academy of Pediatrics estableció pautas para los centros de oncología pediátrica y su función en el tratamiento de los pacientes con cáncer infantil.[2] En estos centros de oncología pediátrica, se dispone de ensayos clínicos para la mayoría de los tipos de cáncer que se presentan en niños y adolescentes, y se ofrece la oportunidad de participar a la mayoría de los pacientes y familiares. Por lo general, los ensayos clínicos para los niños y adolescentes con cáncer se diseñan a fin de comparar un tratamiento que parece mejor con el tratamiento estándar actual. La mayoría de los avances en la identificación de tratamientos curativos para los cánceres infantiles se lograron mediante ensayos clínicos. Para obtener información sobre los ensayos clínicos en curso, consultar el portal de Internet del NCI.
Se han logrado mejoras muy importantes en la supervivencia de niños y adolescentes con cáncer.[3,4,5] Entre 1975 y 2020, la mortalidad por cáncer infantil disminuyó en más de un 50 %.[3,6,7] Los niños y adolescentes supervivientes de cáncer requieren un estrecho seguimiento porque los efectos secundarios de la terapia oncológica pueden persistir o desarrollarse meses o años después del tratamiento. Para obtener información específica sobre la incidencia, el tipo y el seguimiento de los efectos tardíos en niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Referencias:
Smith MA, Seibel NL, Altekruse SF, et al.: Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 28 (15): 2625-34, 2010.
American Academy of Pediatrics: Standards for pediatric cancer centers. Pediatrics 134 (2): 410-4, 2014. Also available online. Last accessed August 23, 2024.
Smith MA, Altekruse SF, Adamson PC, et al.: Declining childhood and adolescent cancer mortality. Cancer 120 (16): 2497-506, 2014.
Childhood cancer. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 28. Also available online. Last accessed August 21, 2023.
Childhood cancer by the ICCC. In: Howlader N, Noone AM, Krapcho M, et al., eds.: SEER Cancer Statistics Review, 1975-2010. National Cancer Institute, 2013, Section 29. Also available online. Last accessed August 21, 2023.
National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed August 23, 2024.
Surveillance Research Program, National Cancer Institute: SEER*Explorer: An interactive website for SEER cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed September 5, 2024.
Tratamiento del retinoblastoma intraocular
Tratamiento del retinoblastoma intraocular unilateral
Las opciones de tratamiento del retinoblastoma intraocular unilateral son las siguientes:
Enucleación para tumores intraoculares grandes, con o sin quimioterapia adyuvante según el riesgo patológico, cuando no es posible salvar el ojo.
Abordajes conservadores de rescate ocular cuando es posible salvar el ojo y la vista:
Quimiorreducción con quimioterapia sistémica o quimioterapia intraarterial y quimioterapia intravítrea o sin esta.
Tratamientos locales, incluso crioterapia, termoterapia y radioterapia con placas.
Enucleación con quimioterapia adyuvante o sin esta
Debido a que la enfermedad unilateral por lo habitual es maciza y, a menudo, no hay ninguna expectativa de que se pueda conservar una visión útil, por lo general, se realiza una cirugía (enucleación) al inicio del tratamiento. Es necesario que el examen meticuloso de la pieza de enucleación lo haga un patólogo con experiencia para determinar la presencia de características de riesgo alto de enfermedad metastásica. Estas características de riesgo alto incluyen las siguientes:[1,2,3,4,5]; [6][Nivel de evidencia C1 y C2]
Diseminación a la cámara anterior.
Compromiso coroideo masivo.
Tumor que se extiende más allá de la lámina cribosa.
Diseminación escleral y extraescleral.
Las imágenes por resonancia magnética antes de la enucleación tienen sensibilidad y especificidad bajas para detectar un tipo patológico de riesgo alto.[7]
El tipo patológico de riesgo alto se ha relacionado con la presencia de diseminación mínima en la médula ósea y el líquido cefalorraquídeo detectada mediante reacción en cadena de la polimerasa cuantitativa para la detección de CRX o de la GD2 sintasa. En un grupo de 96 niños con retinoblastoma no metastásico y características patológicas de riesgo alto, la tasa de supervivencia sin enfermedad a 3 años fue del 78 % en los pacientes con diseminación mínima detectable en comparación con el 98 % en los pacientes sin enfermedad detectable (P = 0,004).[8]
Se ha usado la terapia sistémica adyuvante con vincristina, doxorrubicina y ciclofosfamida, o con vincristina, carboplatino y etopósido para prevenir la enfermedad metastásica en pacientes con ciertas características de riesgo alto evaluadas mediante una revisión patológica después de la enucleación.[3]
En el estudio ARET0332 (NCT00335738) del Children's Oncology Group, se investigó de forma prospectiva la función de la quimioterapia adyuvante en 321 niños aptos con retinoblastoma unilateral enucleado recién diagnosticado. La revisión histopatológica central se realizó en los cortes patológicos de todos los pacientes. Las indicaciones definidas para el uso de la quimioterapia adyuvante fueron el reemplazo masivo de la coroides definido como invasión uveal posterior de grados IIC y IID, cualquier compromiso uveal posterior de menos de 3 mm con compromiso concomitante del nervio óptico y compromiso del nervio óptico posterior a la lámina cribosa. El tratamiento incluyó 6 ciclos de carboplatino, etopósido y vincristina administrado cada 4 semanas.[6][Nivel de evidencia C1 y C2]
La revisión central de los ojos enucleados por patólogos oftálmicos permitió identificar a un grupo de niños (19 % de todos los pacientes aptos) que estaban clasificados por error en el informe patológico institucional.
Se destacó la importancia de directrices universales para manejar e interpretar las muestras de retinoblastoma, y como recurso esencial para el reconocimiento y la categorización de las características de riesgo alto.
Para todos los pacientes del estudio, la tasa general de supervivencia sin complicaciones (SSC) a 2 años fue del 98 % y la tasa de supervivencia general (SG) fue del 99 %.
Para los pacientes con características de riesgo alto que exigieron quimioterapia adyuvante, la tasa de SSC a 2 años fue del 96 %.
Para los pacientes con características de riesgo bajo a quienes se indicó someterse a observación, la tasa de SSC a 2 años fue del 99 %.
La combinación de compromiso extenso de la coroides y el nervio óptico posterior a la lámina cribosa representa el riesgo más alto de recidiva y apoya la necesidad de quimioterapia más intensiva para este grupo.
El tiempo promedio hasta la primera recidiva fue de menos de 12 meses; el tiempo hasta la muerte después de la recidiva fue de cerca de 8 meses.
Abordajes conservadores de rescate ocular
Es posible ofrecer abordajes conservadores de rescate ocular, como la quimioterapia sistémica y los tratamientos de control local, para intentar salvar el ojo y conservar la vista.[9] Las tasas de rescate ocular se correlacionan con el grupo intraocular. La posibilidad de salvar el ojo sin administrar radioterapia de haz externo (RHE) fue superior al 80 % en los niños con enfermedad intraocular en estadio inicial. No obstante, los resultados oculares en los niños con enfermedad intraocular avanzada fueron deficientes cuando se usaron quimioterapia sistémica y tratamientos locales, con tasas de recate ocular inferiores al 40 %, incluso después de la administración de RHE.[10] La braquiterapia con placa se ha usado como terapia de rescate en pacientes con retinoblastoma unilateral. En una serie, 12 ojos de 12 niños se trataron con braquiterapia con placa de rutenio. Se logró el rescate del globo ocular en el 75 % de los pacientes. La tasa de control local final fue del 66 %.[11]
Se debe tener cuidado con el retraso de la enucleación mediante la prolongación del tratamiento con quimioterapia sistémica cuando el control tumoral no parece posible, en particular, en ojos del grupo E. La quimioterapia antes de la enucleación en ojos con enfermedad intraocular avanzada, a veces, conlleva una estadificación inferior y subestimación de los hallazgos patológicos de enfermedad extrarretiniana y extraocular; en consecuencia, aumenta el riesgo de diseminación.[12]
La administración de quimioterapia mediante una cánula en la arteria oftálmica como tratamiento inicial del retinoblastoma unilateral avanzado es más eficaz que la quimiorreducción con quimioterapia sistémica, en especial, para los ojos del grupo D.[13,14]; [15][Nivel de evidencia C3] En un centro multidisciplinario de vanguardia, la quimioterapia intraarterial para el tratamiento de pacientes con retinoblastoma unilateral intraocular avanzado produce tasas de conservación ocular del 70 % al 90 %.[14,15,16,17,18] Para obtener más información, consultar la sección Quimioterapia intraarterial (infusión de quimioterapia en la arteria oftálmica).
La electrorretinografía, una técnica que mide las respuestas eléctricas de varios tipos de células de la retina, como los fotorreceptores, a veces se usa para evaluar el funcionamiento de la retina durante el tratamiento con quimioterapia intraarterial y después de este. En un estudio, la electrorretinografía pretratamiento se correlacionó con la agudeza visual final después de la quimioterapia intraarterial, lo que indica que es posible utilizar esta técnica para ayudar en la toma de decisiones asociadas con las estrategias de tratamiento y la priorización de las intervenciones.[19]
Opciones de tratamiento en evaluación clínica para el retinoblastoma intraocular unilateral
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
ARET2121 (NCT05504291) (A Study to Give Treatment Inside the Eye to Treat Retinoblastoma): en este ensayo se estudia la viabilidad de incorporar inyecciones intravítreas de melfalán durante la quimioterapia sistémica neoadyuvante (carboplatino, vincristina y etopósido). Los pacientes son aptos si tienen retinoblastoma unilateral del grupo D con diseminación vítrea o retinoblastoma bilateral con el ojo peor categorizado como grupo D y diseminación vítrea.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Seguimiento tras el tratamiento del retinoblastoma intraocular unilateral
Debido a que una proporción de niños con retinoblastoma unilateral presentará con el tiempo la enfermedad en el ojo opuesto, estos niños se someten a asesoramiento genético y pruebas genéticas, así como a exámenes periódicos del ojo no afectado, con independencia del tratamiento que reciben. La enfermedad bilateral asincrónica se presenta con mayor frecuencia en pacientes con padres afectados y en niños cuyo diagnóstico se hizo en los primeros meses de vida.
Tratamiento del retinoblastoma bilateral intraocular
El objetivo del tratamiento del retinoblastoma bilateral es conservar el ojo y la vista, así como retardar o evitar la RHE y la enucleación.
Las opciones de tratamiento del retinoblastoma intraocular bilateral son las siguientes:
Enucleación para tumores intraoculares grandes, seguida de quimioterapia adaptada al riesgo y según las características patológicas cuando no es posible salvar el ojo ni la vista.
Abordajes conservadores de rescate ocular cuando es posible salvar el ojo y la vista.
Quimiorreducción con quimioterapia sistémica o quimioterapia intraarterial y quimioterapia intravítrea o sin esta.
Tratamientos locales, incluso crioterapia, termoterapia y radioterapia con placas.
Radioterapia de haz externo.
En general, la carga de tumor intraocular es asimétrica y el tratamiento se determina según el ojo con la enfermedad más avanzada. El tratamiento sistémico se suele seleccionar a partir del ojo con enfermedad más extensa. En los pacientes con enfermedad bilateral, las opciones de tratamiento descritas para la enfermedad unilateral se utilizan en uno o ambos ojos afectados. Si bien es posible que se necesite la enucleación inicial de un ojo con enfermedad avanzada y la administración de quimioterapia adyuvante adaptada al riesgo, es usual que el tratamiento de elección sea un abordaje más conservador que incluya la administración de quimiorreducción primaria y tratamiento local intensivo con seguimiento minucioso de la respuesta. En la actualidad, la RHE se reserva para los pacientes cuyos ojos no reaccionan bien a la quimioterapia sistémica o intraarterial inicial, y a la consolidación local.[20]
Varios centros grandes publicaron los resultados de ensayos en los que se usó quimioterapia sistémica junto con una consolidación local intensiva en pacientes con enfermedad bilateral.[21] Los fármacos de base para la quimiorreducción por lo general son carboplatino, etopósido y vincristina. Aunque la combinación menos tóxica de vincristina y carboplatino logra controlar la enfermedad en una proporción significativa de los pacientes, el rescate ocular es superior cuando el régimen incluye etopósido.[22][Nivel de evidencia B1] Se obtuvieron resultados similares en un estudio de una institución en el que se usó topotecán en lugar de etopósido en un régimen de combinación.[23][Nivel de evidencia C1] Cuando se usa quimiorreducción y control local intensivo, se comprobó que el sistema de agrupamiento de la International Classification of Retinoblastoma predice la supervivencia ocular, con tasas de rescate del globo ocular que, por lo general, superan el 80 % en los grupos A y B, y del 40 % al 80 % en los grupos C y D, aunque quizás se necesite RHE en los casos de enfermedad intraocular avanzada.[24]; [21,23][Nivel de evidencia C1]
La administración de quimioterapia mediante canulación de la arteria oftálmica con adición de quimioterapia intravítrea para pacientes con enfermedad persistente vítrea o subretiniana se volvió una alternativa muy importante al uso de quimioterapia sistémica.[13,14,16,17]; [15][Nivel de evidencia C3] Si bien la administración en tándem es factible, la administración bilateral aumenta el riesgo de efectos tóxicos sistémicos causados por la exposición al melfalán.[25] En estos casos, es posible utilizar quimioterapia intraarterial con carboplatino en monoterapia para tratar el ojo con enfermedad menos avanzada durante el procedimiento en tándem.[26] Estos tratamientos solo se deben administrar en centros oncológicos con infraestructura de vanguardia para el tratamiento y que cuenten con un equipo multidisciplinario especializado. Para obtener más información, consultar las secciones Quimioterapia intraarterial (infusión de quimioterapia en la arteria oftálmica) y Quimioterapia intravítrea.
Para los pacientes que presentan gran carga tumoral intraocular con diseminación subretiniana o vítrea (ojos del grupo D), se exploró la administración de dosis más altas de carboplatino y carboplatino debajo de la cápsula de Tenon, además de dosis más bajas de RHE (36 Gy) para los pacientes con enfermedad persistente. Con este abordaje intensivo, la supervivencia ocular se acerca a una tasa del 70 % a los 60 meses.[27][Nivel de evidencia B4]
El pronóstico de los pacientes con ojos del grupo E tratados con quimioterapia sistémica y medidas de control local es muy precario sin la radioterapia.[27][Nivel de evidencia B4] El uso prolongado de quimioterapia sistémica para ojos del grupo E con la intención de evitar o diferir la enucleación se relacionó con una menor supervivencia específica de la enfermedad.[12][Nivel de evidencia C1]
Opciones de tratamiento en evaluación clínica para el retinoblastoma intraocular bilateral
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación, se presenta un ejemplo de un ensayo clínico nacional o institucional en curso:
ARET2121 (NCT05504291) (A Study to Give Treatment Inside the Eye to Treat Retinoblastoma): en este ensayo se estudia la viabilidad de incorporar inyecciones intravítreas de melfalán durante la quimioterapia sistémica neoadyuvante (carboplatino, vincristina y etopósido). Los pacientes son aptos si tienen retinoblastoma unilateral del grupo D con diseminación vítrea o retinoblastoma bilateral con el ojo peor categorizado como grupo D y diseminación vítrea.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Chantada GL, Guitter MR, Fandiño AC, et al.: Treatment results in patients with retinoblastoma and invasion to the cut end of the optic nerve. Pediatr Blood Cancer 52 (2): 218-22, 2009.
Eagle RC: High-risk features and tumor differentiation in retinoblastoma: a retrospective histopathologic study. Arch Pathol Lab Med 133 (8): 1203-9, 2009.
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
Kaliki S, Shields CL, Rojanaporn D, et al.: High-risk retinoblastoma based on international classification of retinoblastoma: analysis of 519 enucleated eyes. Ophthalmology 120 (5): 997-1003, 2013.
Sastre X, Chantada GL, Doz F, et al.: Proceedings of the consensus meetings from the International Retinoblastoma Staging Working Group on the pathology guidelines for the examination of enucleated eyes and evaluation of prognostic risk factors in retinoblastoma. Arch Pathol Lab Med 133 (8): 1199-202, 2009.
Chévez-Barrios P, Eagle RC, Krailo M, et al.: Study of Unilateral Retinoblastoma With and Without Histopathologic High-Risk Features and the Role of Adjuvant Chemotherapy: A Children's Oncology Group Study. J Clin Oncol 37 (31): 2883-2891, 2019.
Chawla B, Sharma S, Sen S, et al.: Correlation between clinical features, magnetic resonance imaging, and histopathologic findings in retinoblastoma: a prospective study. Ophthalmology 119 (4): 850-6, 2012.
Laurent VE, Torbidoni AV, Sampor C, et al.: Minimal Disseminated Disease in Nonmetastatic Retinoblastoma With High-Risk Pathologic Features and Association With Disease-Free Survival. JAMA Ophthalmol 134 (12): 1374-1379, 2016.
Shields CL, Honavar SG, Meadows AT, et al.: Chemoreduction plus focal therapy for retinoblastoma: factors predictive of need for treatment with external beam radiotherapy or enucleation. Am J Ophthalmol 133 (5): 657-64, 2002.
Shields CL, Honavar SG, Meadows AT, et al.: Chemoreduction for unilateral retinoblastoma. Arch Ophthalmol 120 (12): 1653-8, 2002.
Negretti GS, Quhill H, Duncan C, et al.: Ruthenium plaque radiotherapy in the current era of retinoblastoma treatment. Ophthalmic Genet 43 (6): 756-761, 2022.
Zhao J, Dimaras H, Massey C, et al.: Pre-enucleation chemotherapy for eyes severely affected by retinoblastoma masks risk of tumor extension and increases death from metastasis. J Clin Oncol 29 (7): 845-51, 2011.
Abramson DH, Fabius AW, Issa R, et al.: Advanced Unilateral Retinoblastoma: The Impact of Ophthalmic Artery Chemosurgery on Enucleation Rate and Patient Survival at MSKCC. PLoS One 10 (12): e0145436, 2015.
Munier FL, Mosimann P, Puccinelli F, et al.: First-line intra-arterial versus intravenous chemotherapy in unilateral sporadic group D retinoblastoma: evidence of better visual outcomes, ocular survival and shorter time to success with intra-arterial delivery from retrospective review of 20 years of treatment. Br J Ophthalmol 101 (8): 1086-1093, 2017.
Shields CL, Jorge R, Say EA, et al.: Unilateral Retinoblastoma Managed With Intravenous Chemotherapy Versus Intra-Arterial Chemotherapy. Outcomes Based on the International Classification of Retinoblastoma. Asia Pac J Ophthalmol (Phila) 5 (2): 97-103, 2016 Mar-Apr.
Francis JH, Iyer S, Gobin YP, et al.: Retinoblastoma Vitreous Seed Clouds (Class 3): A Comparison of Treatment with Ophthalmic Artery Chemosurgery with or without Intravitreous and Periocular Chemotherapy. Ophthalmology 124 (10): 1548-1555, 2017.
Francis JH, Levin AM, Zabor EC, et al.: Ten-year experience with ophthalmic artery chemosurgery: Ocular and recurrence-free survival. PLoS One 13 (5): e0197081, 2018.
Wen X, Fan J, Jin M, et al.: Intravenous versus super-selected intra-arterial chemotherapy in children with advanced unilateral retinoblastoma: an open-label, multicentre, randomised trial. Lancet Child Adolesc Health 7 (9): 613-620, 2023.
Levin AM, Francis JH, McFadden M, et al.: Association of electroretinography with visual outcomes after ophthalmic artery chemosurgery for retinoblastoma in ICRb D and E eyes. PLoS One 14 (1): e0210647, 2019.
Orman A, Koru-Sengul T, Miao F, et al.: The modern role of radiation therapy in treating advanced-stage retinoblastoma: long-term outcomes and racial differences. Int J Radiat Oncol Biol Phys 90 (5): 1037-43, 2014.
Rodriguez-Galindo C, Orbach DB, VanderVeen D: Retinoblastoma. Pediatr Clin North Am 62 (1): 201-23, 2015.
Lumbroso-Le Rouic L, Aerts I, Hajage D, et al.: Conservative treatment of retinoblastoma: a prospective phase II randomized trial of neoadjuvant chemotherapy followed by local treatments and chemothermotherapy. Eye (Lond) 30 (1): 46-52, 2016.
Brennan RC, Qaddoumi I, Mao S, et al.: Ocular Salvage and Vision Preservation Using a Topotecan-Based Regimen for Advanced Intraocular Retinoblastoma. J Clin Oncol 35 (1): 72-77, 2017.
Shields CL, Mashayekhi A, Au AK, et al.: The International Classification of Retinoblastoma predicts chemoreduction success. Ophthalmology 113 (12): 2276-80, 2006.
Schaiquevich P, Buitrago E, Taich P, et al.: Pharmacokinetic analysis of melphalan after superselective ophthalmic artery infusion in preclinical models and retinoblastoma patients. Invest Ophthalmol Vis Sci 53 (7): 4205-12, 2012.
Francis JH, Gobin YP, Brodie SE, et al.: Experience of intra-arterial chemosurgery with single agent carboplatin for retinoblastoma. Br J Ophthalmol 96 (9): 1270-1, 2012.
Berry JL, Jubran R, Kim JW, et al.: Long-term outcomes of Group D eyes in bilateral retinoblastoma patients treated with chemoreduction and low-dose IMRT salvage. Pediatr Blood Cancer 60 (4): 688-93, 2013.
Tratamiento del retinoblastoma extraocular
En países de ingresos altos, pocos pacientes de retinoblastoma presentan durante el cuadro clínico inicial una enfermedad extraocular. La enfermedad extraocular a veces se encuentra en los tejidos blandos alrededor del ojo o en el nervio óptico más allá del margen de resección. Sin embargo, es posible que la enfermedad se disemine al encéfalo y las meninges, con diseminación subsiguiente al líquido cefalorraquídeo, así como enfermedad metastásica a distancia que compromete pulmones, huesos y médula ósea.
Tratamiento del retinoblastoma orbitario y locorregional
El retinoblastoma orbitario se presenta como resultado de la progresión tumoral a través de los vasos emisarios y la esclerótica. Por esta razón, la enfermedad transescleral se considera extraocular y se deberá tratar como tal. El retinoblastoma orbitario se presenta en forma aislada en el 60 % al 70 % de los casos.
Las opciones de tratamiento del retinoblastoma extraocular (orbitario y locorregional) son las siguientes:
Quimioterapia.
Radioterapia.
Enucleación (para diseminación extraocular).
El tratamiento incluye quimioterapia sistémica y radioterapia. Este abordaje de tratamiento logra curación en un 60 % a 85 % de los pacientes. Debido a que la mayoría de las recidivas se presentan en el sistema nervioso central (SNC), se recomiendan los regímenes con fármacos de penetración comprobada en el SNC.
El Children's Oncology Group (COG) realizó un ensayo prospectivo internacional (ARET0321 [NCT00554788]) en el que se incluyó a pacientes con retinoblastoma extraocular. El estudio demostró que la terapia intensificada mejoraba los desenlaces de los pacientes con enfermedad en estadio II, III o IVa, en comparación con los controles históricos. Sin embargo, los pacientes en estadio IVb necesitan una terapia más eficaz.[1][Nivel de evidencia B4]
Los pacientes con retinoblastoma en estadio II o III (n = 19) recibieron 4 ciclos de quimioterapia (vincristina, cisplatino, ciclofosfamida, etopósido) seguidos de radioterapia del campo afectado (45 Gy).
La mediana de seguimiento fue de 7,3 años.
Las tasas de supervivencia sin complicaciones (SSC) y de supervivencia general (SG) a 3 años fueron del 88,1 %.
Los pacientes con retinoblastoma en estadio IVa (n = 18) recibieron 4 ciclos de terapia de inducción (mencionada más arriba). Los pacientes con más de una respuesta parcial se sometieron a apoyo con células madre hematopoyéticas autógenas. Los pacientes con tumor residual tras la quimioterapia recibieron radioterapia.
Las tasas de SSC y SG a 3 años fueron del 76,7 %.
Los pacientes con retinoblastoma en estadio IVb (n = 20) recibieron el mismo tratamiento que los pacientes con enfermedad en estadio IVa.
La tasa de SSC a 1 año fue del 28,3 % y la de SG del 42,1 %.
La tasa de SSC a 3 años fue del 14,2 % y la de SG del 12,3 %.
De los pacientes, 6 presentaron neoplasias malignas secundarias. Hubo 1 caso de osteosarcoma (8 años después del tratamiento), 1 caso de carcinoma papilar tiroideo (9 años después del tratamiento), 1 caso de glioblastoma multiforme (8 años después del tratamiento), un caso de trastorno linfoproliferativo de células B grandes relacionado con el virus de Epstein-Barr (8 meses después del tratamiento), 1 caso de leucemia mieloide aguda (2 años después del tratamiento) y 1 caso de sarcoma alveolar de partes blandas de la vejiga (5 años después del tratamiento). De los pacientes, 5 tenían retinoblastoma unilateral, 2 de los cuales recibieron radioterapia (uno unilateral y otro bilateral).
En los pacientes con enfermedad orbitaria macroscópica, es eficaz diferir la cirugía hasta obtener una respuesta quimioterapéutica (por lo habitual, después de 2 o 3 tres ciclos de tratamiento). Luego, los pacientes se someten a enucleación y reciben 4 a 6 cursos de quimioterapia. Durante la consolidación, el paciente recibe terapias de control local con irradiación orbitaria (40–45 Gy). Con este abordaje, no se indica la exenteración orbitaria.[2]
Se considera que los pacientes con compromiso aislado del nervio óptico en el punto de la transección tienen enfermedad extraocular y se tratan con terapia sistémica, similar a la utilizada para la enfermedad orbitaria macroscópica, e irradiación dirigida a toda la órbita (36 Gy) con un refuerzo de 10 Gy de irradiación dirigida al quiasma (total 46 Gy).[3]
Tratamiento de la enfermedad en el sistema nervioso central
La diseminación intracraneal se presenta por diseminación directa a través del nervio óptico. El pronóstico de estos pacientes es desalentador. El tratamiento incluye quimioterapia sistémica intensiva a base de derivados del platino y terapia dirigida al SNC. Aunque tradicionalmente se usó la quimioterapia intratecal, no hay evidencia preclínica o clínica que respalde su uso.
Las opciones de tratamiento del retinoblastoma extraocular (enfermedad en el SNC) son las siguientes:
Quimioterapia sistémica y terapia dirigida al SNC con radioterapia.
Quimioterapia sistémica seguida de quimioterapia mielosupresora, rescate de células madre y radioterapia, o sin esta.
La administración de radioterapia en estos pacientes es polémica. Se han observado respuestas con radiación craneoespinal de 25 a 35 Gy dirigida a todo el eje encefalomedular y un refuerzo (10 Gy) dirigido a los sitios con enfermedad medible.[1]
El COG realizó un estudio prospectivo (ARET0321 [NCT00554788]) de pacientes con retinoblastoma extraocular, en el que se incluyó a pacientes con enfermedad en estadio IVb que fueron tratados con 4 ciclos de terapia de inducción (vincristina, cisplatino, ciclofosfamida y etopósido).[1][Nivel de evidencia B4]
Los pacientes que obtuvieron una respuesta superior a la parcial fueron tratados con carboplatino, tiotepa y etopósido con dosis altas, con apoyo de células madre hematopoyéticas autógenas.
Los pacientes con enfermedad residual tras la quimioterapia recibieron radioterapia.
En los 20 pacientes con enfermedad en estadio IVb, la tasa de SSC a 1 año fue del 28,3 % y la tasa de SG fue del 42,1 %. La tasa de SSC a 3 años fue del 14,2 % y la de SG del 12,3 %.
Tratamiento del retinoblastoma trilateral sincrónico
El retinoblastoma trilateral por lo general se relaciona con una lesión pineal o, en menor frecuencia, con una lesión supraselar.[4,5,6] En pacientes con la forma hereditaria de retinoblastoma, es menos probable que la enfermedad en el SNC sea el resultado de la diseminación metastásica o regional en comparación con un foco intracraneal primario, como un tumor pineal. El pronóstico para los pacientes con retinoblastoma trilateral es muy precario. La mayoría de los pacientes mueren en menos de 9 meses por enfermedad diseminada al sistema nervioso central.[7,8] Sin embargo, el aumento de la vigilancia y la administración de tratamiento intensivo produjo una mejora de la supervivencia del 6 % (pacientes tratados antes de 1995) al 44 % (pacientes tratados después de 1996).[9]
Las opciones de tratamiento del retinoblastoma trilateral sincrónico son las siguientes:
Quimioterapia sistémica seguida de cirugía y quimioterapia mielosupresora con rescate de células madre.
Quimioterapia sistémica seguida de cirugía y radioterapia.
Si bien los pineoblastomas que se presentan en pacientes de más edad son sensibles a la radioterapia, las estrategias actuales se dirigen a evitar la radiación mediante el uso de quimioterapia intensiva seguida de consolidación con quimioterapia mielosupresora y rescate autógeno de células madre hematopoyéticas. Este es un abordaje similar al utilizado para tratar los tumores de encéfalo en lactantes.[10]
Para obtener más información sobre el retinoblastoma trilateral, y los exámenes de detección con neuroimágenes, consultar la sección Retinoblastoma trilateral.
Tratamiento del retinoblastoma extracraneal metastásico
Las opciones de tratamiento del retinoblastoma extracraneal metastásico son las siguientes:
Quimioterapia sistémica seguida de quimioterapia mielosupresora con rescate de células madre y radioterapia.
A veces, se presentan metástasis hematógenas en los huesos, la médula ósea y, con menos frecuencia, en el hígado. El COG realizó un ensayo prospectivo internacional (ARET0321 [NCT00554788]) para pacientes con retinoblastoma extraocular. En el estudio se demostró que la terapia intensificada mejoraba los desenlaces de los pacientes con enfermedad en estadio IVa.[1][Nivel de evidencia B4] Los pacientes con retinoblastoma en estadio IVa (n = 18) fueron tratados con 4 ciclos de terapia de inducción (vincristina, cisplatino, ciclofosfamida y etopósido).
Los pacientes con una respuesta superior a la parcial recibieron un ciclo de carboplatino, tiotepa y etopósido con dosis altas con apoyo de células madre hematopoyéticas autógenas. Las pacientes con tumor residual tras la quimioterapia recibieron radioterapia.
Con una mediana de seguimiento de 7,3 años, las tasas de SSC y SG a 3 años fueron del 76,7 %.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Dunkel IJ, Piao J, Chantada GL, et al.: Intensive Multimodality Therapy for Extraocular Retinoblastoma: A Children's Oncology Group Trial (ARET0321). J Clin Oncol 40 (33): 3839-3847, 2022.
Radhakrishnan V, Kashyap S, Pushker N, et al.: Outcome, pathologic findings, and compliance in orbital retinoblastoma (International Retinoblastoma Staging System stage III) treated with neoadjuvant chemotherapy: a prospective study. Ophthalmology 119 (7): 1470-7, 2012.
Aerts I, Sastre-Garau X, Savignoni A, et al.: Results of a multicenter prospective study on the postoperative treatment of unilateral retinoblastoma after primary enucleation. J Clin Oncol 31 (11): 1458-63, 2013.
Rodjan F, de Graaf P, Brisse HJ, et al.: Trilateral retinoblastoma: neuroimaging characteristics and value of routine brain screening on admission. J Neurooncol 109 (3): 535-44, 2012.
Paulino AC: Trilateral retinoblastoma: is the location of the intracranial tumor important? Cancer 86 (1): 135-41, 1999.
Blach LE, McCormick B, Abramson DH, et al.: Trilateral retinoblastoma--incidence and outcome: a decade of experience. Int J Radiat Oncol Biol Phys 29 (4): 729-33, 1994.
Kivelä T: Trilateral retinoblastoma: a meta-analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol 17 (6): 1829-37, 1999.
Marcus DM, Brooks SE, Leff G, et al.: Trilateral retinoblastoma: insights into histogenesis and management. Surv Ophthalmol 43 (1): 59-70, 1998 Jul-Aug.
de Jong MC, Kors WA, de Graaf P, et al.: Trilateral retinoblastoma: a systematic review and meta-analysis. Lancet Oncol 15 (10): 1157-67, 2014.
Dunkel IJ, Jubran RF, Gururangan S, et al.: Trilateral retinoblastoma: potentially curable with intensive chemotherapy. Pediatr Blood Cancer 54 (3): 384-7, 2010.
Tratamiento del retinoblastoma progresivo o recidivante
El pronóstico de un paciente con retinoblastoma progresivo o recidivante depende de su ubicación y del grado de progresión o recidiva, así como del tratamiento previo.
La introducción de la quimioterapia intravenosa para el tratamiento del retinoblastoma a principios de la década de 1990 revolucionó el abordaje del retinoblastoma. En una revisión retrospectiva de 869 ojos en 551 pacientes con retinoblastoma que se trataron con quimiorreducción, el 64 % de los ojos presentaron una recidiva y el 94 % de las recidivas o de los tumores nuevos se detectaron dentro de los primeros 3 años de tratamiento. Los factores de riesgo para la recidiva fueron los siguientes:[1][Nivel de evidencia C1]
Una edad más joven del paciente en el momento del diagnóstico (oportunidad relativa [OR], 1,02 por 1 mes de disminución; P = 0,02).
Un grupo más avanzado según la International Classification of Retinoblastoma (OR, 1,24 por 1 grupo más avanzado; P = 0,01).
Una menor distancia del tumor a la papila óptica (OR, 1,11 por 1 mm de disminución; P = 0,03).
La presencia de semillas subretinianas (OR, 1,66; P = 0,02).
Las variantes patogénicas germinales contribuyeron al riesgo de formación de nuevos tumores.
Las recidivas intra y extraoculares tienen pronósticos muy diferentes y se tratan de diferentes maneras.
Tratamiento del retinoblastoma intraocular progresivo o recidivante
Las opciones de tratamiento del retinoblastoma intraocular progresivo o recidivante son las siguientes:
Enucleación.
Radioterapia (radioterapia de haz externo o con placas).
Tratamientos locales (crioterapia o termoterapia).
Quimioterapia de rescate (quimioterapia sistémica o intraarterial).
Quimioterapia intravítrea, en especial para la diseminación vítrea resistente al tratamiento o recidivante.
Para obtener más información sobre el uso y las posibles aplicaciones de la quimioterapia intraarterial e intravítrea, consultar las secciones Quimioterapia intraarterial (infusión de quimioterapia en la arteria oftálmica) y Quimioterapia intravítrea.
Es posible que los pacientes con la forma hereditaria de la enfermedad, cuyos ojos se trataron solo con medidas de control local, presenten tumores intraoculares nuevos porque todas las células de la retina tienen una variante del gen RB1. Este evento no se debe considerar recidivante. La vigilancia quizá permita detectar tumores nuevos en un estadio temprano incluso en pacientes muy jóvenes con retinoblastoma hereditario sometidos a tratamiento previo con quimiorreducción e intervenciones de control local. La terapia adicional de control local, incluso la braquiterapia con placas a veces es exitosa para erradicar los tumores.[2]
Cuando la recidiva o la progresión del retinoblastoma son pequeñas y se limitan al ojo, es posible que el pronóstico para la vista y la supervivencia sea excelente solo con terapia local.[3][Nivel de evidencia C3] Si la recidiva o la progresión se limitan al ojo pero son extensas, el pronóstico para la vista es precario; no obstante, la supervivencia continúa siendo excelente.
La quimioterapia intraarterial en la arteria oftálmica ha resultado eficaz en pacientes que recaen después de recibir quimioterapia sistémica y radioterapia.[4,5] La quimioterapia intraarterial de rescate, por lo general con otros fármacos, se ha usado también después de la quimioterapia intraarterial primaria.[6] La radioterapia con placas es una opción para los pacientes que presentan una recidiva de retinoblastoma tras el tratamiento con quimioterapia intraarterial.[7][Nivel de evidencia C3] Se debe considerar la administración de radioterapia en los pacientes que no la recibieron antes. Por último, quizá se necesite la enucleación en casos de enfermedad progresiva luego del fracaso de todos los tratamientos de rescate ocular.
Tratamiento del retinoblastoma extraocular progresivo o recidivante
Las opciones de tratamiento del retinoblastoma extraocular progresivo o recidivante son las siguientes:
Quimioterapia sistémica y radioterapia para la enfermedad orbitaria.
Quimioterapia sistémica seguida de quimioterapia mielosupresora con rescate de células madre y radioterapia para la enfermedad extraorbitaria.
La recidiva en la órbita después de una enucleación se trata con quimioterapia intensiva además de radioterapia local debido al riesgo elevado de enfermedad metastásica.[8][Nivel de evidencia C1] Después de una enucleación por recidiva, las imágenes de resonancia magnética de alta resolución con bobinas orbitarias son útiles para diferenciar una recidiva orbitaria de un realce posquirúrgico.[9]
Si la recidiva o la progresión son extraoculares, la probabilidad de supervivencia es baja.[10] Sin embargo, es posible que la administración de quimioterapia sistémica intensiva y la consolidación con dosis altas de quimioterapia y rescate autógeno de células madre hematopoyéticas mejore la probabilidad de cura; en especial, en los pacientes con recidiva extracraneal. Se debe considerar la participación en ensayos clínicos de los pacientes cuya enfermedad recidivó después de que se sometieran a estos abordajes intensivos. Para obtener más información, consultar la sección Tratamiento del retinoblastoma extraocular.
Opciones de tratamiento en evaluación clínica para el retinoblastoma progresivo o recidivante
Un abordaje en investigación para los pacientes con retinoblastoma intraocular progresivo incluye el uso de un adenovirus oncolítico que se dirige a RB1.[11]
La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Dalvin LA, Bas Z, Tadepalli S, et al.: Risk Factors for Tumor Recurrence Following Primary Intravenous Chemotherapy (Chemoreduction) for Retinoblastoma in 869 Eyes of 551 Patients. J Pediatr Ophthalmol Strabismus 57 (4): 224-234, 2020.
Wilson MW, Haik BG, Billups CA, et al.: Incidence of new tumor formation in patients with hereditary retinoblastoma treated with primary systemic chemotherapy: is there a preventive effect? Ophthalmology 114 (11): 2077-82, 2007.
Chan MP, Hungerford JL, Kingston JE, et al.: Salvage external beam radiotherapy after failed primary chemotherapy for bilateral retinoblastoma: rate of eye and vision preservation. Br J Ophthalmol 93 (7): 891-4, 2009.
Schaiquevich P, Ceciliano A, Millan N, et al.: Intra-arterial chemotherapy is more effective than sequential periocular and intravenous chemotherapy as salvage treatment for relapsed retinoblastoma. Pediatr Blood Cancer 60 (5): 766-70, 2013.
Say EA, Iyer PG, Hasanreisoglu M, et al.: Secondary and tertiary intra-arterial chemotherapy for massive persistent or recurrent subretinal retinoblastoma seeds following previous chemotherapy exposure: long-term tumor control and globe salvage in 30 eyes. J AAPOS 20 (4): 337-42, 2016.
Francis JH, Abramson DH, Gobin YP, et al.: Efficacy and toxicity of second-course ophthalmic artery chemosurgery for retinoblastoma. Ophthalmology 122 (5): 1016-22, 2015.
Ruben M, Eiger-Moscovich M, Yaghy A, et al.: Iodine-125 Plaque Radiotherapy for Retinoblastoma Recurrence Following Intra-arterial Chemotherapy. J Pediatr Ophthalmol Strabismus 59 (3): 164-171, 2022 May-Jun.
Kim JW, Kathpalia V, Dunkel IJ, et al.: Orbital recurrence of retinoblastoma following enucleation. Br J Ophthalmol 93 (4): 463-7, 2009.
Sirin S, de Jong MC, de Graaf P, et al.: High-Resolution Magnetic Resonance Imaging Can Reliably Detect Orbital Tumor Recurrence after Enucleation in Children with Retinoblastoma. Ophthalmology 123 (3): 635-45, 2016.
Broaddus E, Topham A, Singh AD: Survival with retinoblastoma in the USA: 1975-2004. Br J Ophthalmol 93 (1): 24-7, 2009.
Pascual-Pasto G, Bazan-Peregrino M, Olaciregui NG, et al.: Therapeutic targeting of the RB1 pathway in retinoblastoma with the oncolytic adenovirus VCN-01. Sci Transl Med 11 (476): , 2019.
Efectos tardíos del tratamiento del retinoblastoma
En un informe del Retinoblastoma Survivor Study (N = 470), el 87 % de los sobrevivientes de retinoblastoma (mediana de edad, 43 años; mediana de seguimiento, 42 años) presentaron al menos una afección y el 71 % padecieron una afección grave o potencialmente mortal. En comparación con los pacientes sin retinoblastoma, el riesgo relativo ajustado de una afección crónica en los sobrevivientes fue de 1,4 (P < 0,01). El riesgo relativo de una afección de grado 3 o 4 fue de 7,6 (P < 0,01). Después de excluir las afecciones oculares y las neoplasias subsiguientes (NS), se encontró que este riesgo excesivo solo persistió en los pacientes con enfermedad bilateral.[1]
Neoplasias subsiguientes
La muerte por neoplasias subsiguientes (NS) es la causa de muerte más común en los pacientes de retinoblastoma. Las NS explican cerca del 50 % de las defunciones de pacientes con enfermedad bilateral y retinoblastoma hereditario definido a partir del análisis genético.[2,3,4] Los sobrevivientes de retinoblastoma tienen un riesgo alto de presentar NS.
Los factores que afectan el riesgo de NS son los siguientes:
Retinoblastoma hereditario. Los pacientes con retinoblastoma hereditario tienen un aumento marcado de la incidencia de NS, que es independiente del tratamiento con radioterapia.[2,3,5] En una serie alemana de 643 pacientes con retinoblastoma hereditario, la quimioterapia con radioterapia, o sin esta, fue el único factor de riesgo significativo de inicio de segundos cánceres fuera de la región periorbitaria.[3] Es posible que haya una relación entre el tipo de variante de RB1 y la incidencia de NS; la pérdida completa de la actividad de RB1 se relaciona con una incidencia más alta de NS.[6] Los pacientes heterocigotos para variantes de RB1 con penetrancia regular quizás exhiban un riesgo más alto de presentar NS que los pacientes con variantes de RB1 con penetrancia incompleta.[7] La penetrancia regular se refiere a los pacientes con enfermedad hereditaria y tumores bilaterales multifocales clásicos. La penetrancia incompleta, aunque rara, es un fenotipo clínico en el que los familiares afectados quizás no presentan retinoblastoma ni exhiban una enfermedad unilateral. Con el aumento de la supervivencia de pacientes de retinoblastoma hereditario, se ha hecho evidente que estos pacientes también están en riesgo de cánceres epiteliales posteriores durante la edad adulta. Se ha descrito un gran aumento de la mortalidad por cáncer de pulmón, vejiga y otros cánceres epiteliales.[8,9]
En una serie numerosa de dos instituciones, se identificaron 2053 pacientes con retinoblastoma (diagnosticado entre 1914 y 2016) con un seguimiento máximo de 70 años. La mayoría de las muertes se presentaron en pacientes con retinoblastoma hereditario (518 de 1129) y las NS causaron 267 de esas muertes. Se notificó aumento en el riesgo de muerte por cánceres de páncreas, intestino grueso y riñón. El riesgo general de NS fue más alto para los pacientes tratados con radioterapia y quimioterapia en comparación con los pacientes tratados con radioterapia sola, aunque la distribución varió según el órgano. En una cohorte de 143 sobrevivientes de retinoblastoma diagnosticado entre 1997 y 2006, se observó una mejora continua de la mortalidad.[4][Nivel de evidencia C1] En pacientes con retinoblastoma no hereditario, solo 27 muertes entre 924 pacientes se atribuyeron a NS.
En los sobrevivientes de retinoblastoma hereditario, aquellos con una variante patogénica germinal heredada tienen un riesgo un poco más alto de NS que quienes exhiben una variante de novo. El melanoma fue la NS más común en los pacientes con variantes patogénicas germinales.[10]
Antecedentes de tratamiento con radioterapia para el retinoblastoma. Se notificó que la incidencia acumulada de NS es del 26 % (± 10 %) en pacientes no irradiados y del 58 % (± 10 %) en pacientes irradiados 50 años después del diagnóstico de retinoblastoma, que resulta en una tasa anual de alrededor del 1 % por año.[11]
En una serie alemana de 633 pacientes con retinoblastoma hereditario se observó una tasa de supervivencia a 5 años del 93 %. No obstante, 40 años después, solo el 80 % de los pacientes sobrevivieron y la mayoría padeció NS causadas por radiación (cociente de riesgos instantáneos, alrededor de 3).[12] En otros estudios en los que se analizaron cohortes de pacientes tratados con planificación y técnicas de administración de radiación más avanzadas, se notificaron tasas de NS de alrededor del 9,4 % en pacientes no irradiados y del 30,4 % en pacientes irradiados.[13]
En un estudio no aleatorizado en el que se compararon 2 cohortes contemporáneas de pacientes con retinoblastoma hereditario tratados con terapia con fotones (n = 31) o terapia con protones (n = 55), la incidencia acumulada a 10 años de NS fue significativamente diferente en los 2 grupos (0 % para la radiación con protones vs. 14 % para la radiación con fotones, P = 0,015).[14] Se necesita un seguimiento más prolongado para definir mejor el riesgo de NS relacionadas con la radiación con protones.
La NS más común es el sarcoma, en especial, el osteosarcoma, seguido del sarcoma de tejido blando y el melanoma. Estas neoplasias malignas se presentan dentro y fuera del campo de radiación, aunque en su mayoría son inducidas por la radiación. El efecto cancerígeno de la radioterapia se relaciona con la dosis administrada, en particular, para sarcomas subsiguientes. El aumento por etapas es claro en todas las categorías de dosis. En pacientes irradiados, dos tercios de las NS surgen dentro del tejido irradiado y un tercio, fuera del campo de radiación.[5,11,13,15]
En una cohorte de 952 sobrevivientes de retinoblastoma hereditario irradiados que fueron diagnosticados originalmente entre 1914 y 2006, se identificaron 105 sarcomas óseos y 125 sarcomas de tejido blando. Aproximadamente dos tercios de estos cánceres se presentaron en la cabeza y el cuello. Las tasas de incidencia fueron 2000 veces más altas para los sarcomas óseos y 500 veces más altas para los sarcomas de tejido blando que lo que se esperaba en la población general. Los sarcomas óseos de cabeza y cuello y de tejido blando se diagnosticaron en la primera infancia y continuaron hasta la edad adulta, con una incidencia acumulada a 60 años del 6,8 % para los sarcomas óseos y del 9,3 % para los sarcomas de tejido blando. Los sarcomas óseos y de tejido blando diagnosticados en otras partes del cuerpo aumentaron 169 y 45,7 veces, respectivamente, en comparación con la población general. Los sarcomas óseos se presentaron principalmente en los huesos largos durante la adolescencia. La incidencia de sarcomas de tejido blando fue rara hasta los 30 años, cuando aumentó abruptamente (incidencia acumulada a 60 años, 6,6 %), en particular en mujeres (9,4 %). Los sarcomas de tejido blando en mujeres fueron leiomiosarcomas y se localizaron principalmente en el abdomen y la pelvis.[5,11]; [16][Nivel de evidencia C1]
Edad en el momento de la radioterapia. Al parecer el riesgo de NS depende de la edad del paciente en el momento en que se administra la radioterapia de haz externo (RHE); en particular, en niños menores de 12 meses. Además, la edad parece afectar los tipos histopatológicos de NS.[13,17,18]
NS previas. Los sobrevivientes de una NS tienen un riesgo 7 veces más alto de presentar otra NS.[19] El riesgo aumenta 3 veces más para los pacientes que reciben radioterapia.[20]
No se ha resuelto el problema de equilibrar el control tumoral a largo plazo y las consecuencias de la quimioterapia. La mayoría de los pacientes que reciben quimioterapia se exponen a etopósido, que se ha relacionado con leucemia secundaria en pacientes sin predisposición al cáncer. Sin embargo, la mayoría de los pacientes están expuestos a tasas moderadas en comparación con los riesgos relacionados con la RHE para el retinoblastoma hereditario.
A pesar de que se conoce el aumento del riesgo de leucemia mieloide aguda (LMA) relacionado con el etopósido, los pacientes con retinoblastoma hereditario no exhiben un riesgo alto de presentar esta NS.[21,22,23] En un informe inicial con métodos de encuestas informales, se describió a 15 pacientes que presentaron LMA después de la quimioterapia. La mitad de los pacientes también recibió radioterapia.[22] Este hallazgo no se corroboró en estudios formales. En un estudio de una sola institución con 245 pacientes que recibieron etopósido, solo 1 paciente presentó leucemia promielocítica aguda después de 79 meses.[21] Además, en el Surveillance, Epidemiology, and End Result Registry (SEER) Program se calcularon las tasas de incidencia estandarizadas de neoplasias malignas hematopoyéticas secundarias en 34 867 sobrevivientes de cáncer infantil. El cociente entre la incidencia observada y la prevista para la LMA secundaria en pacientes que recibieron tratamiento para retinoblastoma fue de 0.[24]
La supervivencia después de presentar NS es definitivamente subóptima y varía mucho entre los estudios.[8,25,26,27,28,29] Sin embargo, con los avances terapéuticos, es fundamental que todas las NS en los sobrevivientes de retinoblastoma se traten con intención curativa.[30]
Otros efectos tardíos
Otros efectos tardíos que a veces se presentan después del tratamiento del retinoblastoma son los siguientes:
Disminución del crecimiento orbitario. El crecimiento orbitario disminuye un poco después de la enucleación. Sin embargo, el efecto de la enucleación en el volumen orbitario quizás se minimice después de la colocación de un implante orbitario.[31] La asimetría en el tamaño de la órbita que se produce después de la enucleación es mayor en los niños pequeños y en los lactantes, pero la enucleación después de los 3 años no produce asimetría orbitaria.[32]
Deficiencia de campos visuales. Los pacientes de retinoblastoma presentan una variedad de defectos visuales a largo plazo después del tratamiento de la enfermedad intraocular. Estos defectos se relacionan con el tamaño del tumor, la localización y el método de tratamiento.[33]
Se realizó un estudio de agudeza visual después del tratamiento con quimioterapia sistémica y terapia oftálmica local en 54 ojos de 40 niños. Al cabo de una media de seguimiento de 68 meses, 27 ojos (50 %) tuvieron una agudeza visual final de 20/40 o superior, y 36 ojos (67 %) una agudeza visual final de 20/200 o superior. Los factores clínicos que permitieron pronosticar una agudeza visual de 20/40 o superior fueron un margen tumoral de por lo menos 3 mm desde la fovéola y la papila óptica, y la ausencia de líquido subretiniano.[34]
Estrabismo. En un estudio de 42 sobrevivientes de retinoblastoma (64 ojos) sometidos a un tratamiento conservador exitoso, se observó que el 69 % de los pacientes presentaban estrabismo al final del seguimiento (media, 93,7 meses). El estrabismo divergente fue el tipo de estrabismo más común notificado. Los tratamientos conservadores incluyeron quimioterapia intravenosa (62 ojos), quimioterapia intraarterial (10 ojos), braquiterapia (11 ojos), termoterapia transpupilar (22 ojos), crioterapia (47 ojos) y radioterapia de haz externo (4 ojos). En el análisis multivariante, solo el compromiso de la fóvea resultó ser un factor de riesgo significativo para presentar estrabismo (P < 0,001).[35]
Hipoacusia. Debido a que el carboplatino sistémico se usa con regularidad en el tratamiento del retinoblastoma, surgió una preocupación sobre la hipoacusia relacionada con el tratamiento. Para obtener más información sobre el tratamiento, consultar las secciones Tratamiento del retinoblastoma intraocular y Tratamiento del retinoblastoma extraocular.
Pese a que en 2 estudios numerosos de niños sometidos a 6 ciclos de terapia con carboplatino (18,6 mg/kg por ciclo), se observó una incidencia de hipoacusia relacionada con el tratamiento menor del 1 %,[36,37] En una serie diferente se documentó algún grado de hipoacusia en el 17 % de los pacientes.[38] En este último estudio, una edad menor (menos de 6 meses en el momento del tratamiento) y la exposición a dosis más altas de carboplatino sistémico se relacionaron con un mayor riesgo de ototoxicidad.[38,39]
Funcionamiento neurocognitivo. En los primeros estudios sobre el funcionamiento intelectual en sobrevivientes de retinoblastoma se indicó una inteligencia superior al promedio entre los sobrevivientes de enfermedad bilateral en comparación con los hermanos no afectados y la población general, en especial en aquellos que quedaron ciegos por su enfermedad.[40,41,42]
En estudios posteriores los resultados han sido heterogéneos con hallazgos contradictorios, en parte, como resultado de la baja fiabilidad de prueba y repetición de las medidas usadas para evaluar los desenlaces cognitivos a una edad muy temprana, así como por las diferencias temporales en la exposición al tratamiento.
La evaluación en serie del funcionamiento cognitivo y adaptativo en un grupo de sobrevivientes menores de 6 años reveló disminuciones en el funcionamiento del desarrollo a lo largo del tiempo; las más pronunciadas se observaron en pacientes con deleciones 13q.[43]
Durante el seguimiento longitudinal posterior de esta cohorte se identificaron mejoras del funcionamiento adaptativo en todos los grupos de tratamiento y mejoras del funcionamiento cognitivo en los sobrevivientes que se trataron con enucleación sola entre los 5 y los 10 años de edad.[44]
A los 10 años de edad, el funcionamiento general estaba por lo común dentro del intervalo promedio, aunque el coeficiente intelectual era significativamente más bajo que la media normativa de los niños que se trataron con enucleación sola.[44]
En un estudio de sobrevivientes adultos de retinoblastoma a muy largo plazo, con una media de 33 años desde el diagnóstico, se demostró un funcionamiento cognitivo en gran medida dentro del promedio para las competencias de inteligencia, memoria, atención y función ejecutiva.[45]
Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en los niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Referencias:
Friedman DN, Chou JF, Oeffinger KC, et al.: Chronic medical conditions in adult survivors of retinoblastoma: Results of the Retinoblastoma Survivor Study. Cancer 122 (5): 773-81, 2016.
Shinohara ET, DeWees T, Perkins SM: Subsequent malignancies and their effect on survival in patients with retinoblastoma. Pediatr Blood Cancer 61 (1): 116-9, 2014.
Temming P, Arendt M, Viehmann A, et al.: Incidence of second cancers after radiotherapy and systemic chemotherapy in heritable retinoblastoma survivors: A report from the German reference center. Pediatr Blood Cancer 64 (1): 71-80, 2017.
Kleinerman RA, Tucker MA, Sigel BS, et al.: Patterns of Cause-Specific Mortality Among 2053 Survivors of Retinoblastoma, 1914-2016. J Natl Cancer Inst 111 (9): 961-969, 2019.
MacCarthy A, Bayne AM, Brownbill PA, et al.: Second and subsequent tumours among 1927 retinoblastoma patients diagnosed in Britain 1951-2004. Br J Cancer 108 (12): 2455-63, 2013.
Dommering CJ, Marees T, van der Hout AH, et al.: RB1 mutations and second primary malignancies after hereditary retinoblastoma. Fam Cancer 11 (2): 225-33, 2012.
Ketteler P, Hülsenbeck I, Frank M, et al.: The impact of RB1 genotype on incidence of second tumours in heritable retinoblastoma. Eur J Cancer 133: 47-55, 2020.
Fletcher O, Easton D, Anderson K, et al.: Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst 96 (5): 357-63, 2004.
Marees T, van Leeuwen FE, de Boer MR, et al.: Cancer mortality in long-term survivors of retinoblastoma. Eur J Cancer 45 (18): 3245-53, 2009.
Kleinerman RA, Yu CL, Little MP, et al.: Variation of second cancer risk by family history of retinoblastoma among long-term survivors. J Clin Oncol 30 (9): 950-7, 2012.
Wong FL, Boice JD, Abramson DH, et al.: Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. JAMA 278 (15): 1262-7, 1997.
Temming P, Arendt M, Viehmann A, et al.: How Eye-Preserving Therapy Affects Long-Term Overall Survival in Heritable Retinoblastoma Survivors. J Clin Oncol 34 (26): 3183-8, 2016.
Kleinerman RA, Tucker MA, Tarone RE, et al.: Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: an extended follow-up. J Clin Oncol 23 (10): 2272-9, 2005.
Sethi RV, Shih HA, Yeap BY, et al.: Second nonocular tumors among survivors of retinoblastoma treated with contemporary photon and proton radiotherapy. Cancer 120 (1): 126-33, 2014.
Kleinerman RA, Tucker MA, Abramson DH, et al.: Risk of soft tissue sarcomas by individual subtype in survivors of hereditary retinoblastoma. J Natl Cancer Inst 99 (1): 24-31, 2007.
Kleinerman RA, Schonfeld SJ, Sigel BS, et al.: Bone and Soft-Tissue Sarcoma Risk in Long-Term Survivors of Hereditary Retinoblastoma Treated With Radiation. J Clin Oncol 37 (35): 3436-3445, 2019.
Abramson DH, Frank CM: Second nonocular tumors in survivors of bilateral retinoblastoma: a possible age effect on radiation-related risk. Ophthalmology 105 (4): 573-9; discussion 579-80, 1998.
Moll AC, Imhof SM, Schouten-Van Meeteren AY, et al.: Second primary tumors in hereditary retinoblastoma: a register-based study, 1945-1997: is there an age effect on radiation-related risk? Ophthalmology 108 (6): 1109-14, 2001.
Abramson DH, Melson MR, Dunkel IJ, et al.: Third (fourth and fifth) nonocular tumors in survivors of retinoblastoma. Ophthalmology 108 (10): 1868-76, 2001.
Marees T, van Leeuwen FE, Schaapveld M, et al.: Risk of third malignancies and death after a second malignancy in retinoblastoma survivors. Eur J Cancer 46 (11): 2052-8, 2010.
Turaka K, Shields CL, Meadows AT, et al.: Second malignant neoplasms following chemoreduction with carboplatin, etoposide, and vincristine in 245 patients with intraocular retinoblastoma. Pediatr Blood Cancer 59 (1): 121-5, 2012.
Gombos DS, Hungerford J, Abramson DH, et al.: Secondary acute myelogenous leukemia in patients with retinoblastoma: is chemotherapy a factor? Ophthalmology 114 (7): 1378-83, 2007.
Tamboli D, Topham A, Singh N, et al.: Retinoblastoma: A SEER Dataset Evaluation for Treatment Patterns, Survival, and Second Malignant Neoplasms. Am J Ophthalmol 160 (5): 953-8, 2015.
Rihani R, Bazzeh F, Faqih N, et al.: Secondary hematopoietic malignancies in survivors of childhood cancer: an analysis of 111 cases from the Surveillance, Epidemiology, and End Result-9 registry. Cancer 116 (18): 4385-94, 2010.
Yu CL, Tucker MA, Abramson DH, et al.: Cause-specific mortality in long-term survivors of retinoblastoma. J Natl Cancer Inst 101 (8): 581-91, 2009.
Aerts I, Pacquement H, Doz F, et al.: Outcome of second malignancies after retinoblastoma: a retrospective analysis of 25 patients treated at the Institut Curie. Eur J Cancer 40 (10): 1522-9, 2004.
Eng C, Li FP, Abramson DH, et al.: Mortality from second tumors among long-term survivors of retinoblastoma. J Natl Cancer Inst 85 (14): 1121-8, 1993.
Dunkel IJ, Gerald WL, Rosenfield NS, et al.: Outcome of patients with a history of bilateral retinoblastoma treated for a second malignancy: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 30 (1): 59-62, 1998.
Marees T, Moll AC, Imhof SM, et al.: Risk of second malignancies in survivors of retinoblastoma: more than 40 years of follow-up. J Natl Cancer Inst 100 (24): 1771-9, 2008.
Moll AC, Imhof SM, Bouter LM, et al.: Second primary tumors in patients with retinoblastoma. A review of the literature. Ophthalmic Genet 18 (1): 27-34, 1997.
Chojniak MM, Chojniak R, Testa ML, et al.: Abnormal orbital growth in children submitted to enucleation for retinoblastoma treatment. J Pediatr Hematol Oncol 34 (3): e102-5, 2012.
Oatts JT, Robbins JA, de Alba Campomanes AG: The effect of enucleation on orbital growth in patients with retinoblastoma. J AAPOS 21 (4): 309-312, 2017.
Demirci H, Shields CL, Meadows AT, et al.: Long-term visual outcome following chemoreduction for retinoblastoma. Arch Ophthalmol 123 (11): 1525-30, 2005.
Fabian ID, Stacey AW, Naeem Z, et al.: Strabismus in retinoblastoma survivors with long-term follow-up. J AAPOS 22 (4): 276.e1-276.e7, 2018.
Lambert MP, Shields C, Meadows AT: A retrospective review of hearing in children with retinoblastoma treated with carboplatin-based chemotherapy. Pediatr Blood Cancer 50 (2): 223-6, 2008.
Batra A, Thakar A, Bakhshi S: Ototoxicity in retinoblastoma survivors treated with carboplatin based chemotherapy: A cross-sectional study of 116 patients. Pediatr Blood Cancer 62 (11): 2060, 2015.
Qaddoumi I, Bass JK, Wu J, et al.: Carboplatin-associated ototoxicity in children with retinoblastoma. J Clin Oncol 30 (10): 1034-41, 2012.
Leahey A: A cautionary tale: dosing chemotherapy in infants with retinoblastoma. J Clin Oncol 30 (10): 1023-4, 2012.
Levitt EA, Rosenbaum AL, Willerman L, et al.: Intelligence of retinoblastoma patients and their siblings. Child Dev 43 (3): 939-48, 1972.
Eldridge R, O'Meara K, Kitchin D: Superior intelligence in sighted retinoblastoma patients and their families. J Med Genet 9 (3): 331-5, 1972.
Williams M: Superior intelligence of children blinded from retinoblastoma. Arch Dis Child 43 (228): 204-10, 1968.
Willard VW, Qaddoumi I, Chen S, et al.: Developmental and adaptive functioning in children with retinoblastoma: a longitudinal investigation. J Clin Oncol 32 (25): 2788-93, 2014.
Willard VW, Qaddoumi I, Pan H, et al.: Cognitive and Adaptive Functioning in Youth With Retinoblastoma: A Longitudinal Investigation Through 10 Years of Age. J Clin Oncol 39 (24): 2676-2684, 2021.
Brinkman TM, Merchant TE, Li Z, et al.: Cognitive function and social attainment in adult survivors of retinoblastoma: a report from the St. Jude Lifetime Cohort Study. Cancer 121 (1): 123-31, 2015.
Actualizaciones más recientes a este resumen (09 / 19 / 2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes hechos a este resumen a partir de la fecha arriba indicada.
Se modificó el formato de este resumen.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del retinoblastoma. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del retinoblastoma son:
Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
Thomas A. Olson, MD (Aflac Cancer and Blood Disorders Center of Children's Healthcare of Atlanta - Egleston Campus)
D. Williams Parsons, MD, PhD (Texas Children's Hospital)
Carlos Rodriguez-Galindo, MD (St. Jude Children's Research Hospital)
Nita Louise Seibel, MD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.