Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Resumen
Esta sección es una síntesis de los temas tratados en este resumen del PDQ sobre las características genéticas del cáncer de piel; incluye enlaces a las secciones específicas que tratan la evidencia de cada tema.
- Herencia y riesgo
Más de 100 tipos de tumores exhiben manifestaciones clínicas en la piel. Muchos de estos tumores tienen un componente familiar o hereditario, ya sea aislado o como parte de un síndrome con otras manifestaciones. El carcinoma de células basales (CCB) y el carcinoma de células escamosas (CCE) son dos de las neoplasias malignas más frecuentes en los Estados Unidos. La causa suele ser la exposición solar, aunque hay varios genes y síndromes hereditarios asociados con aumento del riesgo de estas neoplasias. El melanoma de piel (que también se conoce como melanoma cutáneo) es un tipo menos común de cáncer de piel, pero un 5 % a un 10 % de todos los melanomas surgen en familias con casos múltiples y se puede heredar de manera autosómica dominante. El melanoma es el cáncer más mortal de los tipos comunes de cáncer de piel.
- Genes y síndromes asociados
Varios genes y síndromes hereditarios se asocian con la aparición del cáncer de piel, por ejemplo, los siguientes:
- Carcinoma de células basales. El síndrome de nevo de células basales (SNCB, causado por variantes patogénicas de PTCH1 y en algunos casos por variantes de PTCH2) se asocia con riesgo elevado de CCB.
- Carcinoma de células escamosas. Los síndromes como el albinismo oculocutáneo, la epidermolisis bullosa y la anemia de Fanconi se asocian con riesgo elevado de CCE.
- Melanoma. El gen CDKN2A es un importante gen supresor de tumores de la línea germinal que se asocia con riesgo elevado de melanoma. Las variantes patogénicas de CDKN2A explican entre el 35 % al 40 % de todos los melanomas familiares. Hay variantes germinales patogénicas de otros genes (por ejemplo, CDK4, MITF y BAP1) que también se vinculan con riesgo elevado de melanoma.
Una enfermedad autosómica recesiva, llamada xeroderma pigmentoso (XP), se asocia con un aumento del riesgo de presentar CCB, CCE y melanoma.
Los estudios de asociación del genoma completo son prometedores para identificar alelos de susceptibilidad de penetrancia baja que son comunes para muchas enfermedades complejas, como el melanoma; pero no se ha determinado la utilidad clínica de estos hallazgos.
- Cuidado clínico
Las estrategias de reducción del riesgo para las personas con predisposición hereditaria al cáncer de piel son semejantes a las recomendaciones para la población general. Estas recomendaciones abarcan evitar la exposición al sol, aplicar protector solar, usar ropa de protección solar y evitar el uso de las cámaras de bronceado. Se han estudiado fármacos de quimioprevención como la isotretinoína y la acitretina para el tratamiento de los CCE en pacientes con SNCB y XP; estos producen una disminución significativa en el número de tumores por año. El vismodegib también demostró ser promisorio para la reducción de la tasa anual por paciente de casos nuevos de CCB que requieren tratamiento quirúrgico en personas con SNCB. También se observó que la isotretinoína reduce la incidencia de CCE en pacientes con XP.
El tratamiento del cáncer de piel hereditario es similar al tratamiento del cáncer de piel esporádico. En un estudio de una población con XP, se encontró que el uso terapéutico de fluorouracilo (5-FU) es eficaz, en especial, para el tratamiento de lesiones extensas. Además de su función terapéutica y como posible fármaco de quimioprevención, el vismodegib también está en estudio para determinar posibles efectos paliativos en tumores odontogénicos queratoquísticos de pacientes con SNCB.
- Aspectos psicosociales y del comportamiento
La mayor parte de la bibliografía sobre los aspectos psicosociales de los tipos de cáncer de piel hereditarios se ha centrado en los pacientes con melanoma familiar. En las personas con riesgo de melanoma familiar, los factores psicosociales repercuten en las decisiones sobre la toma de pruebas genéticas para determinar el riesgo de cáncer hereditario y las estrategias de manejo del riesgo. Por lo general, el interés en las pruebas genéticas para las variantes patogénicas del gen CDKN2A es alto. Los beneficios percibidos en las personas con una historia familiar significativa de melanoma incluyen la información sobre el riesgo de melanoma personal y para la descendencia, además del incremento en la motivación para poner en práctica conductas de protección solar. En varios estudios se examinaron conductas de reducción del riesgo y detección temprana en personas con historia familiar de melanoma. En general, estos estudios indican una adopción y mantenimiento irregulares de estos comportamientos. Los estudios de intervención se han orientado al conocimiento sobre el melanoma, la protección solar y las conductas de detección en familiares de pacientes con melanoma, pero los resultados son contradictorios. Se están llevando a cabo investigaciones para comprender y abordar mejor los problemas psicosociales y de comportamiento en las familias de riesgo alto.
Introducción
Estructura de la piel
La genética del cáncer de piel es un tema demasiado amplio. Más de 100 tipos de tumores exhiben manifestaciones clínicas en la piel, muchos de estos tumores tienen un componente familiar, ya sea aislado o como parte de un síndrome que se acompaña de otras manifestaciones. Esto se debe, en parte, a que la piel en sí es un órgano complejo compuesto de varios tipos de células. Además, muchos de estos tipos de células pueden exhibir una transformación maligna en varios puntos de su diferenciación, lo que conduce a tumores con características histológicas específicas y comportamientos biológicos muy diferentes, como el carcinoma de células escamosas (CCE) y el cáncer de células basales (CCB). Estos tipos de cáncer se conocen en conjunto como cánceres de piel no melanoma o cánceres de queratinocitos.
La Figura 1 es una ilustración sencilla de la estructura de la piel normal. También se observan los principales tipos de células que se encuentran en cada compartimiento. En términos generales, hay dos compartimentos grandes —la epidermis avascular y la dermis vascular— con muchos tipos de células distribuidos dentro de una matriz de tejido conjuntivo formada casi en su totalidad por fibroblastos.[1]
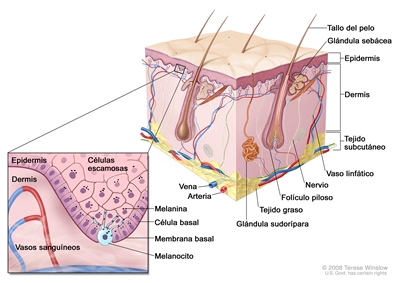
La capa externa o epidermis está compuesta sobre todo por queratinocitos, pero también contiene varios tipos de poblaciones celulares minoritarias. La capa más interna está compuesta de queratinocitos basales contiguos a la membrana basal y melanocitos entremezclados. La membrana basal es una estructura importante desde el punto de vista anatómico y funcional, que contiene productos derivados de los queratinocitos y de los fibroblastos cutáneos, como colágeno y laminina. Los queratinocitos basales van perdiendo contacto con la membrana basal al dividirse. A medida que los queratinocitos basales migran hacia la superficie de la piel, exhiben una diferenciación progresiva, pierden el núcleo y forman la capa de células espinosas, la capa de células granulosas y la capa externa queratinizada, o estrato córneo, que sirve como cubierta protectora del cuerpo.
El verdadero origen citológico del CCB sigue sin estar claro. Los queratinocitos basales y las células del CCB comparten muchas similitudes histológicas, como lo refleja su nombre. De manera alternativa, también se ha planteado que el origen del CCB son las células de la vaina radicular externa del folículo piloso.[2] Este planteamiento se fundamenta en el hecho de que el CCB ocurre de manera predominante en la piel con folículos pilosos. Los CCB casi nunca metastatizan, pero a veces invaden tejidos locales o regionales, y en ocasiones se propagan por los nervios.[3]
Todavía se debate el origen del CCE; no obstante, es probable que este tipo de cáncer se derive de las células madre epidérmicas asociadas con el folículo piloso.[4] El CCE puede surgir en diversos tejidos, como los del pulmón y el cuello uterino, lo que determina un comportamiento un poco diferente según el tejido de origen. Incluso en el cáncer derivado de la piel, el CCE exhibe diferentes grados de malignidad según el sitio de la piel donde aparece; por ejemplo, el CCE de áreas lampiñas (lisas, sin vello) de la piel expuesta al sol tiene una tasa metastásica más baja que el CCE que surge en el borde libre de los labios o en cicatrices.[3]
Además, dentro del compartimento epidérmico, los melanocitos se distribuyen de manera individual a lo largo de la membrana basal y a veces sufren una transformación maligna a un melanoma. Los melanocitos se derivan de las células de la cresta neural y migran al compartimento epidérmico hacia la 8.a semana de edad gestacional. Los melanocitos contienen melanina, que se almacena en melanosomas y se transporta a los queratinocitos cercanos para inducir pigmentación en la piel. La melanina proporciona un barrera que protege el núcleo de los queratinocitos contra la radiación ultravioleta, además cumple una función en el sistema inmunitario.[5]
Las células de Langerhans o células dendríticas, son otro tipo de célula de la epidermis cuya función principal es la presentación de antígenos. Estas células permanecen en la piel durante periodos prolongados y responden a diferentes estímulos, como la radiación ultravioleta o los corticoesteroides tópicos, que estimulan su migración fuera de la piel.[6]
La dermis se compone en gran medida de una matriz extracelular. Las células y orgánulos predominantes en este compartimiento son los fibroblastos, las células endoteliales, las células del músculo liso, las células transitorias del sistema inmunitario, los vasos sanguíneos y los nervios. Cuando se presenta una transformación maligna, los fibroblastos forman fibrosarcomas y las células endoteliales forman angiosarcomas, sarcomas de Kaposi y otros tumores vasculares. Varios tipos de células inmunitarias entran y salen de la piel por los vasos sanguíneos y linfáticos; entre ellas, mastocitos, linfocitos, células mononucleares, histiocitos y granulocitos. Cuando hay enfermedades inflamatorias es posible que aumente la cantidad de estas células y que formen tumores en la piel. Por ejemplo, la urticaria pigmentosa es una afección que surge de los mastocitos y en ocasiones se relaciona con la leucemia de mastocitos. El linfoma cutáneo de células T a menudo se limita a la piel durante toda su evolución. En general, el 10 % de las leucemias y los linfomas exhiben manifestaciones prominentes en la piel.[7]
En el compartimento dérmico también se encuentran las faneras, anejos cutáneos o anexos de la piel, que se derivan de los queratinocitos epidérmicos, y que abarcan los folículos pilosos, las glándulas sudoríparas, y las glándulas sebáceas de los folículos pilosos. Por lo general, estas estructuras se forman durante el primer o segundo trimestre del desarrollo fetal. En las faneras se pueden originar una gran variedad de tumores benignos o malignos con comportamiento biológico variable. Varios de estos tumores se asocian con síndromes familiares. En conjunto, hay docenas de subtipos histológicos de estos tumores asociados con componentes individuales de los anejos.[8]
Por último, la hipodermis o tejido subcutáneo es una capa que se extiende por debajo de la dermis a una profundidad variable según la ubicación anatómica. El límite más profundo puede corresponder a músculo, fascia, hueso o cartílago. Hay afecciones inflamatorias que afectan la hipodermis, como la paniculitis y algunas neoplasias malignas, como el liposarcoma.[9]
En cada uno de estos compartimientos se originan varias neoplasias malignas propias, pero además comprenden un área de diseminación adyacente inmediata de cánceres de piel localizados que surgen en otros compartimientos. Los límites de cada compartimiento cutáneo se usan para determinar la estadificación de los cánceres de piel. Por ejemplo, un melanoma in situ se limita a la epidermis. Cuando el cáncer cruza la membrana basal y llega a la dermis, se considera un cáncer invasivo. Además, hay neoplasias malignas internas que suelen metastatizar a la piel. La dermis y la hipodermis son los lugares más comunes de metástasis, pero algunas afecciones, como el cáncer de mama Pagetoide, afectan la epidermis.
Funciones de la piel
Las funciones de la piel son muy variadas. En primer lugar, la piel es un barrera importante que previene la pérdida generalizada de agua y calor, y protege contra las abrasiones menores. Cuando se presenta un cáncer es posible que no haya regulación adecuada de estas funciones. Por ejemplo, la eritrodermia (enrojecimiento generalizado de la piel) asociada a una quemadura solar grave altera la regulación de la temperatura corporal, lo que puede conllevar una pérdida importante de calor.
En segundo lugar, la piel tiene importantes funciones en la inmunidad innata y adaptativa. En la inmunidad adaptativa, las células presentadoras de antígenos producen respuestas en los linfocitos T que conllevan un aumento de las concentraciones de linfocitos T auxiliares (TH)1, TH 2 o TH 17.[10] En la inmunidad innata, el sistema inmunitario produce muchos péptidos con efectos antibacterianos y antifúngicos. Incluso las lesiones pequeñas en la piel podrían derivar en una infección. El tejido linfoide de la piel es una de las armas más potentes del sistema inmunitario y cumple una función en la prevención de infecciones. Además, tal vez sea importante en la vigilancia inmunitaria contra el cáncer. La inmunodepresión, como la que se induce de manera intencional después de un trasplante de órgano para reducir el riesgo de rechazo al trasplante, es un factor de riesgo importante para el cáncer de piel. La piel es esencial para la comunicación a través de la expresión facial y los movimientos de las manos. Desafortunadamente, las áreas con función especializada, como el área alrededor de los ojos y los oídos, son lugares comunes de presentación de cáncer. Incluso los cánceres pequeños en estas áreas pueden llevar a desafíos reconstructivos y tienen repercusiones cosméticas y sociales de importancia.[1]
Cuadro clínico inicial de los cánceres de piel
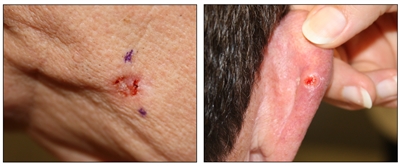
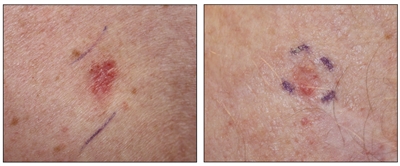
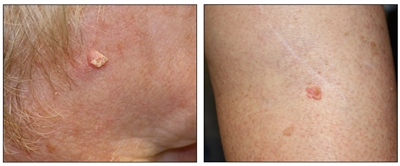
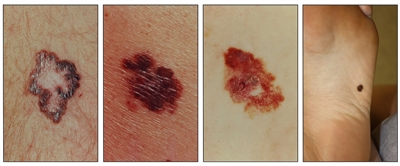
Si bien el aspecto de cada cáncer de piel es variable, hay características físicas generales útiles para la detección sistemática. Los CCB suelen tener un borde nacarado o un aspecto eccematoso (para obtener más información, consultar la Figura 2 y la Figura 3). Además es común que presenten ulceración (para obtener más información, consultar la Figura 2). Los CCE suelen tener una capa gruesa de queratina superficial (para obtener más información, consultar la Figura 4). Tanto los CCB como los CCE están asociados con antecedentes de fotodaño (piel dañada por el sol). Los melanomas se caracterizan por pigmentación oscura y asimétrica, bordes irregulares, variación del color, un diámetro superior a 6 mm y progresión (criterios ABCDE). Para obtener más información en inglés sobre los criterios ABCDE, consultar What Does Melanoma Look Like? en el sitio web del NCI. En las fotografías que siguen se observa la presentación clínica característica de cada uno de estos tipos de cáncer.
Carcinomas de células basales
Carcinomas de células escamosas
Melanomas
Referencias:
- Vandergriff TW, Bergstresser PR: Anatomy and physiology. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 43-54.
- Schirren CG, Rütten A, Kaudewitz P, et al.: Trichoblastoma and basal cell carcinoma are neoplasms with follicular differentiation sharing the same profile of cytokeratin intermediate filaments. Am J Dermatopathol 19 (4): 341-50, 1997.
- Soyer HP, Rigel DS, Wurm EM: Actinic keratosis, basal cell carcinoma and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1773-93.
- Lapouge G, Youssef KK, Vokaer B, et al.: Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A 108 (18): 7431-6, 2011.
- Lin JY, Fisher DE: Melanocyte biology and skin pigmentation. Nature 445 (7130): 843-50, 2007.
- Koster MI, Loomis CA, Koss TK, et al.: Skin development and maintenance. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 55-64.
- Kamino H, Reddy VB, Pui J: Fibrous and fibrohistiocytic proliferations of the skin and tendons. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1961-77.
- McCalmont TH: Adnexal neoplasms. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1829-50.
- Kaddu S, Kohler S: Muscle, adipose and cartilage neoplasms. In: Bolognia JL, Jorizzo JL, Schaffer JV: Dermatology. 3rd ed. Elsevier Saunders, 2012, pp 1979-92.
- Harrington LE, Mangan PR, Weaver CT: Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol 18 (3): 349-56, 2006.
Carcinomas de células basales
Introducción
El carcinoma de células basales (CCB) es la neoplasia maligna más frecuente en personas de ascendencia europea, con un riesgo de por vida del 30 %.[1] Si bien la exposición a la radiación ultravioleta (UV) es el factor de riesgo con un vínculo más estrecho con la formación del CCB, otros factores ambientales (como la radiación ionizante, la ingestión crónica de arsénico y la inmunodepresión) y genéticos (como la historia familiar, el tipo de piel y los síndromes genéticos) también podrían contribuir a la carcinogénesis. En contraste con el melanoma, la diseminación metastásica del CCB es muy infrecuente, y por lo general se origina en tumores grandes que no se han sometido a tratamiento médico durante un período prolongado. Los CCB invaden a nivel local o regional y, en ocasiones, siguen el recorrido de los nervios. El pronóstico del CCB es excelente cuando se detecta temprano.
Factores de riesgo del carcinoma de células basales
Esta sección se centra en los factores de riesgo en personas con predisposición hereditaria al CCB. Para obtener más información sobre los factores de riesgo del CCB en la población general, consultar Prevención del cáncer de piel.
Exposición solar
La exposición al sol es el principal factor ambiental conocido relacionado con el desarrollo de todos los tipos de cáncer de piel. Hay diferentes patrones de exposición al sol que se vinculan con cada uno de los principales tipos de cáncer de piel (CCB, carcinoma de células escamosas [CCE] y melanoma). Para obtener más información, consultar Prevención del cáncer de piel.
Características pigmentarias
El fenotipo de riesgo alto se refiere a personas con las siguientes características físicas:
- Piel clara que se quema con facilidad.
- Iris poco pigmentados (ojos azules o verdes).
- Pecas en la piel expuesta al sol.
- Dificultad para broncearse.
- Cabello rubio o rojo.
En concreto, las personas con piel más pigmentada muestran una incidencia más baja de CCB que las personas con piel poco pigmentada. En un estudio pequeño de casos y controles, las personas con piel de tipo Fitzpatrick I o II (piel más clara) presentaron el doble de riesgo de CCB.[2] El cabello rubio o rojo se vinculó con aumento del riesgo de CCB en 2 grandes cohortes: el Nurses' Health Study y el Health Professionals' Follow-Up Study.[3] En las mujeres del Nurses' Health Study, se observó aumento del riesgo de CCB en aquellas de cabello rojo en comparación con las de cabello castaño claro (riesgo relativo [RR] ajustado, 1,30; intervalo de confianza [IC] 95 %, 1,20–1,40). En los hombres del Health Professionals Follow-Up Study, el riesgo de CCB asociado con el cabello rojo no fue tan alto (RR, 1,17; IC 95 %, 1,02–1,34) y no se consideró significativo después del ajuste por historia familiar de melanoma y antecedentes de quemadura solar.[3] El riesgo relacionado con el cabello rubio fue alto en hombres y mujeres (RR, análisis conjunto, 1,09; IC 95 %, 1,02–1,18), y se consideró que el cabello castaño confería un efecto protector contra el CCB (RR, análisis conjunto, 0,89; IC 95 %, 0,87–0,92). Para obtener más información, consultar la sección Características pigmentarias en la sección Melanoma.
Historia familiar
Las personas con CCB o CCE reportan una frecuencia más alta de estos tipos de cáncer en su familia en comparación con los controles. La importancia de este hallazgo no está clara. Aparte de los trastornos genéticos específicos que confieren un aumento del riesgo de CCB, una historia familiar de cualquier tipo de cáncer de piel es un factor predictivo importante del desarrollo de CCB. Los resultados del Nurses' Health Study y del Health Professionals Follow-Up Study indican que la historia familiar de melanoma en un familiar de primer grado (FPG) se vincula con aumento del riesgo de CCB en hombres y mujeres (RR, 1,31; IC 95 %, 1,25–1,37; P < 0,0001).[3] En las mismas cohortes y en el Nurses' Health Study 2, se observó que la historia familiar de melanoma acarreó un aumento equivalente en el riesgo (cociente de riesgos instantáneos [CRI], 1,27; IC 95 %, 1,12–1,44).[4] En un estudio de 376 casos de CCB de inicio o establecimiento temprano y 383 controles se encontró que la historia familiar de cualquier tipo de cáncer de piel aumentó el riesgo de CCB de inicio temprano (oportunidad relativa [OR], 2,49; IC 95 %, 1,80–3,45). Este riesgo aumentó cuando se diagnosticó cáncer de piel a un FPG antes de los 50 años de edad (OR, 4,79; IC 95 %, 2,90–7,90). Las personas con una historia familiar de melanoma y cáncer de piel no melanoma (NMSC) presentaron el riesgo más alto (OR, 3,65; IC 95 %, 1,79–7,47).[5]
En un estudio sobre la heredabilidad del cáncer en 80 309 gemelos monocigóticos y 123 382 gemelos dicigóticos, se observó que los NMSC tienen una heredabilidad del 43 % (IC 95 %, 26–59 %), lo que indica que casi la mitad del riesgo de este cáncer se debe a factores hereditarios.[6] Además, el riesgo acumulado del NMSC fue 1,9 veces más alto para los gemelos monocigóticos que para los gemelos dicigóticos (IC 95 %, 1,8–2,0).[6]
Antecedentes personales de carcinoma de células basales o de células escamosas
Los antecedentes personales de CCB o CCE se relacionan de manera importante con el desarrollo subsecuente de un CCB o CCE. Hay un aumento aproximado del 20 % en el riesgo de una lesión subsiguiente en el primer año posterior al diagnóstico de un cáncer de piel. La media de edad de aparición de estos tipos de cáncer se ubica alrededor de los 65 años.[7,8,9,10,11,12] Además, en varios estudios se encontró que las personas con antecedentes de CCB o CCE exhiben un riesgo alto (intervalo, 9 %–61 %) de diagnóstico subsiguiente de un cáncer extracutáneo;[13,14,15,16,17,18] sin embargo, otros estudios contradicen estos resultados.[19,20,21,22] En ausencia de otros factores de riesgo o indicios de un síndrome de susceptibilidad al cáncer definido, como se analiza más adelante, en los pacientes con cáncer de piel se fomenta el cumplimiento de las recomendaciones de detección de otros tipos de cáncer extracutáneos orientadas a la población general.
Principales genes asociados con el carcinoma de células basales
PTCH1
Las variantes patogénicas hereditarias del gen que codifica la proteína receptora transmembrana PTCH1 o PTCH se relacionan con el síndrome de nevo de células basales (SNCB), también conocido como síndrome de carcinoma nevoide de células basales o síndrome de Gorlin, mientras que las variantes somáticas de este gen se relacionan con los CCB cutáneos esporádicos. PTCH1, el homólogo humano del gen patched (ptc) de polaridad de segmento de la Drosophila, es un componente integral de la vía de señalización del erizo sónico (hedgehog), que cumple varias funciones durante el desarrollo (formación de las extremidades, segmentación embrionaria, diferenciación del tubo neural) y de regulación (mantenimiento de células madre). Para obtener más información, consultar la sección Síndrome de nevo de células basales.
En el estado de reposo, la proteína receptora transmembrana PTCH1, mediante su actividad catalítica, inhibe la proteína transmembrana 7 Smoothened (Smo), por lo que evita la posterior transducción de la señal.[23] La unión del ligando del erizo sónico con PTCH1 contrarresta el efecto inhibitorio de la Smo, lo que se traduce en la activación de factores de transcripción (GLI1, GLI2), genes de proliferación celular (ciclina D, ciclina E, myc) y reguladores de la angiogénesis.[24,25] Por lo tanto, el equilibrio entre PTCH1 (inhibición) y Smo (activación) controla la secuencia posterior de la vía de transducción de señales del erizo sónico, que es esencial. Las variantes patogénicas de pérdida de función de PTCH1 o las variantes de ganancia de función de Smo inclinan este equilibrio hacia la activación, un acontecimiento clave en la eventual transformación neoplásica.
En el CCB esporádico y en el familiar, la demostración de una pérdida alélica en el cromosoma 9q22 indicó la posibilidad de un gen supresor de tumores.[26,27] En investigaciones posteriores se identificó una variante patogénica del gen PTCH1 que se encontró en el área de la pérdida alélica.[28] Hasta el 30 % de los CCB esporádicos exhiben variantes patogénicas de PTCH1.[29] Otros tumores, además del CCB, el meduloblastoma y el rabdomiosarcoma, se han asociado con variantes patogénicas de PTCH1. Las 3 neoplasias malignas se asocian con SNCB, y la mayoría de las personas con características clínicas de este síndrome presentan variantes germinales patogénicas de PTCH1, con predominio del tipo que produce truncamiento de la proteína.[30]
PTCH2
Se han observado variantes patogénicas truncadoras de PTCH2, un homólogo de PTCH1 ubicado en el cromosoma 1p32.1-32.3, en el CCB y en el meduloblastoma.[31,32]PTCH2 exhibe un 57 % de homología con PTCH1.[33] Aunque no se conoce la función específica de PTCH2, hay indicios que apoyan su participación en la vía de señalización del erizo sónico (hedgehog).[31,34] Sin embargo, se ha cuestionado la función de este gen en el SNCB.[35]
Genes posiblemente asociados con el carcinoma de células basales
BAP1(Proteína 1 asociada a BRCA1)
Las variantes patogénicas del gen BAP1 se relacionan con un aumento del riesgo de varios tipos de cáncer, como el melanoma cutáneo y el melanoma uveal. Aunque no se conoce la penetrancia del CCB en personas con variantes patogénicas BAP1, se han reportado varias familias con variantes de BAP1 con diagnósticos de CCB.[36,37] En un estudio, se reportó el diagnóstico de CCB en personas portadoras de variantes patogénicas en 4 familias. El uso de pruebas inmunohistoquímicas en muestras de tumores demostró la pérdida de la proteína BAP1 en casos de CCB en 2 portadores de variantes patogénicas germinales de BAP1, pero no en 53 casos esporádicos de CCB.[36] En otro estudio se encontró que se diagnosticaron 19 CCB en 4 personas de familias con mutaciones germinales patogénicas de BAP1. Se observó la pérdida completa de la expresión nuclear de BAP1 en 17 de 19 CCB de estas personas, pero no en las muestras de CCB de 22 controles.[38] La pérdida de expresión nuclear de BAP1 también se notificó en una serie de 7 CCB de personas con variantes de pérdida de función de BAP1, pero solo en 1 de 31 CCB esporádicos.[39] Para obtener más información, consultar la sección Proteína 1 asociada a BRCA1 (BAP1) en la sección Melanoma.
MC1R
En un metanálisis se observó que a mayor número de variantes patogénicas de MC1R en el portador, mayor es el riesgo de presentar CCE y CCB. Las personas con 2 o más variantes patogénicas de MC1R presentaron una OR de 2,48 (IC 95 %, 1,96–3,15) para el CCB y una OR de 2,80 (IC 95 %, 1,71–4,57) para el CCE; este riesgo aumentó en personas de cabello rojo.[40] En un estudio de personas con diagnóstico de CCB antes de los 40 años también se encontró una asociación más fuerte entre el CCB y las variantes patogénicas de MC1R en personas con características fenotípicas que no suelen considerarse de riesgo alto. Para obtener más información, consultar la sección MC1R en la sección Melanoma.[41]
Síndromes de predisposición al carcinoma de células basales
Síndrome de nevo de células basales
El síndrome de nevo o nevoide de células basales (SNCB), que también se llama síndrome de Gorlin, síndrome de Gorlin-Goltz y síndrome del CCB nevoide, es una enfermedad autosómica dominante con una prevalencia estimada de 1 en 57 000 personas.[42] Este síndrome se caracteriza por penetrancia completa y niveles elevados de expresividad variable, como lo indica la evaluación de personas con genotipos idénticos, pero fenotipos muy variables.[30,43] Las características clínicas del SNCB varían más entre diferentes familias que dentro de una misma familia.[44] El SNCB se vincula de manera primaria con variantes germinales patogénicas del gen PTCH1, pero hay familias con este fenotipo que exhiben alteraciones en los genes PTCH2 y SUFU.[45,46,47]
Como se detalló antes, PTCH1 participa en el desarrollo y tiene funciones reguladoras; de manera que las variantes germinales patogénicas espontáneas o hereditarias de PTCH1 en el SNCB pueden dar lugar a una variedad amplia de manifestaciones físicas con potencial diagnóstico. La variante patogénica de SNCB se ubica en el cromosoma 9q22.3-q31, y tiene una puntuación del logaritmo de las probabilidades (LOD) máxima de 3,597 y 6,457 en los marcadores D9S12 y D9S53.[42] La consecuente haploinsuficiencia de PTCH1 en el SNCB se asoció con anormalidades estructurales como los queratoquistes odontogénicos, con la evaluación del revestimiento del quiste se demostró pérdida de heterocigosidad (LOH) de PTCH1.[48] Se cree que la formación del CCB y otras neoplasias malignas asociadas con el SNCB siguen el modelo de "doble golpe" (two hit hypothesis) de un gen supresor de tumores: el primer golpe o hit sería la variante germinal patogénica de PTCH1 que produce la heterocigosis inicial, y el segundo golpe sería una mutación en el otro alelo que resulta de la exposición a radiación ionizante o radiación UV.[49,50,51,52,53] Sin embargo, también se ha encontrado que la inactivación de PTCH1 se produce por haploinsuficiencia o isoformas dominantes negativas.[54]
El diagnóstico de SNCB por lo general se basa en hallazgos clínicos y radiológicos característicos. Se usan varios conjuntos de criterios diagnósticos para el SNCB (para obtener más información, consultar el Cuadro 1).[55,56,57,58] Aunque cada conjunto de criterios tienen ventajas y desventajas, ninguno es claramente superior en cuanto al equilibrio entre la sensibilidad y la especificidad para identificar portadores de variantes patogénicas. El SNCB Colloquium Group planteó criterios en 2011 que exigen la presencia de 1 criterio mayor cuando hay pruebas de diagnóstico molecular, mientras que cuando no se han hecho pruebas de diagnóstico molecular se aceptan 2 criterios mayores o 1 criterio mayor acompañado de 2 criterios menores para el diagnóstico de SNCB.[58] Las variantes patogénicas de PTCH1 se encuentran en el 60 % al 85 % de los pacientes que cumplen estos criterios clínicos.[59,60,61] Cabe destacar que el SNCB se relaciona con la formación de neoplasias benignas y malignas. La asociación más fuerte con neoplasias benignas se produce con los fibromas ováricos que se diagnostican en el 14 % al 24 % de las mujeres afectadas por SNCB.[52,56,62] Es más probable que los fibromas de ovario relacionados con el SNCB sean bilaterales y calcificados en comparación con los fibromas ováricos esporádicos.[63] También se ha propuesto la inclusión de los ameloblastomas, tumores agresivos del epitelio odontogénico del maxilar, como un criterio diagnóstico del SNCB, pero la mayoría de grupos todavía no lo incluyen.[64]
Otras neoplasias benignas relacionadas son los pólipos gástricos harmatomatosos,[65] los quistes pulmonares congénitos,[66] los fibromas cardíacos,[67] los meningiomas,[68,69,70] los craneofaringiomas,[71] los rabdomiomas fetales,[72] los leiomiomas,[73] los mesenquimomas,[74] los hamartomas foliculares basaloides,[75] y los tumores dermoides nasales. En la población pediátrica general, se ha documentado la aparición de meningiomas y ependimomas después de radioterapia. Es posible que la radioterapia para procesos intracraneales relacionados con el síndrome sea responsable, en parte, de un subconjunto de estos tumores benignos en personas con SNCB.[76,77,78] Además, es posible que la radioterapia para los meduloblastomas malignos en la población con SNCB cause muchos CCB cutáneos en los campos de radiación. Del mismo modo, el tratamiento del CCB cutáneo con radioterapia puede dar lugar a la inducción de un gran número de CCB adicionales.[51,52,73]
Entre las personas que cumplían con los criterios diagnósticos de SNCB, fue más probable que el diagnóstico se hiciera a una edad más temprana en quienes tenían variantes patogénicas de PTCH1 que en aquellas que no tenían las variantes patogénicas de PTCH1 (19 y 36 años, respectivamente). También fue más probable que estas personas que cumplían con los criterios diagnósticos y tenían las variantes patogénicas de PTCH1 presentaran manifestaciones clínicas como quistes mandibulares (con variantes de PTCH1, 63 %; sin variantes de PTCH1, 34 %), costillas bífidas (con variantes de PTCH1, 56 %; sin variantes de PTCH1, 34 %), o cualquier hallazgo esquelético (con variantes de PTCH1, 74 %; sin variantes de PTCH1, 51 %).[61] También se observaron diferencias entre genotipo y fenotipo. Aquellas personas con variantes de cambio de sentido de PTCH1 tenían una mediana de edad más tardía en el momento del diagnóstico (26 años), menos CCB y menor incidencia de quistes mandibulares que las personas con otros tipos de variantes patogénicas de PTCH1.
Los criterios diagnósticos del SNCB se describen en el Cuadro 1 a continuación.
| Evans et al. 1993[55] | Kimonis et al. 1997[56] | Veenstra-Knol et al. 2005[57] | SNCB Colloquium Group 2011b[58] |
|---|---|---|---|
| CCB = carcinoma de células basales. | |||
| a Se necesitan 2 criterios mayores o 1 criterio mayor acompañado de 2 criterios menores para cumplir con los requisitos para el diagnóstico del SNCB.[55,56,57] | |||
| b El diagnóstico se basa en 1 criterio mayor cuando hay pruebas de diagnóstico molecular, pero cuando no se han hecho pruebas de diagnóstico molecular se aceptan 2 criterios mayores o 1 criterio mayor acompañado de 2 criterios menores.[58] | |||
| Criterios principalesa | |||
| >2 CCB a cualquier edad, 1 CCB diagnosticado antes de los 30 años o >10 nevos de células basales | >2 CCB a cualquier edad o 1 CCB diagnosticado antes de los 20 años | >2 CCB a cualquier edad o 1 CCB diagnosticado antes de los 20 años | Aparición de un CCB antes de los 20 años o un número excesivo de CCB fuera de proporción de acuerdo a la exposición solar previa y el tipo de piel |
| Queratoquiste odontogénico en el maxilar o un quiste óseo poliostótico comprobados por análisis histológico | Queratoquiste odontogénico en el maxilar comprobado por análisis histológico | Queratoquiste odontogénico en el maxilar comprobado por análisis histológico | Queratoquiste odontogénico en el maxilar antes de los 20 años |
| ≥3 hoyuelospalmaresoplantares | ≥3 hoyuelos palmares o plantares | ≥3 hoyuelos palmares o plantares | Hoyuelos palmoplantares |
| Calcificaciones ectópicas, calcificaciones encefálicas laminillares o tempranas (diagnosticadas antes de los 20 años) de la hoz cerebral | Calcificación bilaminar de la hoz cerebral | Calcificación ectópica (laminillar o temprana de la hoz cerebral) encefálicas | Calcificación bilaminar de la hoz cerebral |
| Historia familiar de SNCB | Unfamiliar de primer gradocon SNCB. | Historia familiar de SNCB | Un familiar de primer grado con SNCB. |
| (Las anomalías en las costillas se enumeran como criterio menor; consultar más adelante) | Costillas bífidas, fusionadas o muy separadas | Costillas bífidas, fusionadas o muy separadas | (Las anomalías en las costillas se enumeran como criterio menor; consultar más adelante) |
| (El meduloblastoma es un criterio menor; consultar más adelante) | (El meduloblastoma es un criterio menor; consultar más adelante) | (El meduloblastoma es un criterio menor; consultar más adelante) | Meduloblastoma (usualmente desmoplásico) |
| Criterios menores | |||
| Perímetro occipitofrontal >97 centil y frente prominente | Macrocefalia (ajustada por talla) | Macrocefalia (>97 centil) | Macrocefalia |
| Anomalías esquelética congénitas: costillas bífidas, fusionadas, muy separadas; costillas ausentes o bífidas; vértebras en cuña o fusionadas. | Puente en la silla turca, anomalías vertebrales (hemivértebras, fusión o elongación de cuerpos vertebrales), deformidades de las manos y los pies, radiolucencias con forma de llama en manos y pies. | Puente en la silla turca, anomalías vertebrales (hemivértebras, fusión o elongación de cuerpos vertebrales), deformidades de las manos y los pies | Deformidades esqueléticas (anomalías vertebrales, acortamiento del 4. metacarpiano, polidactilia postaxial) |
| (Las anomalías de las costillas se enumeran como criterio mayor; consultar atrás) | (Las anomalías de las costillas se enumeran como criterio mayor; consultar atrás) | Anomalías en las costillas | |
| Fibroma cardiaco u ovárico | Fibroma ovárico | Fibroma cardiaco u ovárico | Fibroma cardiaco u ovárico |
| Meduloblastoma | Meduloblastoma | Meduloblastoma | (El meduloblastoma es un criterio mayor; consultar atrás) |
| Deformidad congénita: fisura labial o palatina, polidactilia, cataratas, coloboma, microftalmia | Fisura labial o palatina, frente prominente, hipotelorismo moderado o grave | Fisura labial o palatina, polidactilia | Fisura labial o palatina |
| Deformidad de Sprengel, deformidad del tórax marcada, sindactilia pronunciada | Deformidad de Sprengel, deformidad del tórax marcada, sindactilia pronunciada | ||
| Quistes linfomesentéricos | Quistes linfomesentéricos | ||
| Anomalías oculares: cataratas, coloboma, microftalmia | Anomalías oculares (estrabismo, hipotelorismo, cataratas congénitas, coloboma) | ||
Una de las principales preocupaciones en personas con el SNCB, son las neoplasias malignas asociadas, la más común de ellas es el CCB. El CCB en personas con SNCB puede aparecer incluso durante la niñez como lesiones pequeñas similares a los acrocordones; las lesiones más grandes exhiben características cutáneas más clásicas.[79] Los CCB no pigmentados son más comunes que las lesiones pigmentadas.[80] La edad en el momento del primer diagnóstico de CCB asociado al SNCB oscila entre los 3 a 53 años y la media de edad es de 21,4 años. El diagnóstico se establece antes de los 20 años en la gran mayoría de las personas afectadas.[56,62] La mayoría de los CCB se ubican en sitios expuestos al sol, pero las personas que tienen más de 100 CCB exhiben una distribución más uniforme por todo el cuerpo.[80] En series de casos se ha indicado que 1 de cada 200 personas con CCB presentan hallazgos compatibles con el diagnóstico de SNCB.[42] Se han notificado casos de SNCB en personas con pigmentación cutánea más oscura; sin embargo en personas de ascendencia africana o mediterránea se encuentran muchos menos casos de CCB.[56,81,82] A pesar de la escasa frecuencia del CCB en esta población, en los casos reportados se reportó la presencia de todas las manifestaciones extracutáneas del SNCB.[82] Sin embargo, en personas de ascendencia africana que han recibido radioterapia se ha notificado una carga de tumores de células basales significativa alrededor de los campos de radioterapia.[56,73] Por lo tanto, es posible que la pigmentación cutánea confiera protección contra los efectos mutagénicos de la radiación UV, pero no contra los efectos de la radiación ionizante.
Las variantes de otros genes relacionados con un aumento del riesgo del CCB en la población general parecen modificar la edad de aparición del CCB en personas con SNCB. En un estudio de 125 personas con SNCB se encontró que una variante del gen MC1R (Arg151Cys) se relacionó con una mediana de edad de inicio temprana de 27 años (IC 95 %, 20–34), en comparación con las personas que no portaban el alelo de riesgo, quienes presentaron una mediana de edad de inicio del CCB de 34 años (IC 95 %, 30–40) (CRI, 1,64; IC 95 %, 1,04–2,58, P = 0,034). Una variante del gen TERT-CLPTM1L mostró un efecto similar; las personas con el alelo de riesgo presentaron una mediana de edad de inicio del CCB de 31 años (IC 95 %, 28–37) en comparación con una mediana de inicio del CCB de 41 años (IC 95 %, 32–48) en personas que no portaron un alelo de riesgo (CRI, 1,44; IC 95 %, 1,08–1,93, P = 014).[83]
Muchas otras neoplasias malignas se han relacionado con el SNCB. El meduloblastoma presenta la relación más fuerte con el SNCB y se diagnostica en el 1 % al 5 % de los casos de SNCB. Si bien el meduloblastoma asociado al SNCB suele diagnosticarse entre los 2 y 3 años de edad, el meduloblastoma esporádico por lo general se diagnostica entre los 6 y 10 años de edad.[52,56,62,84] El fenotipo desmoplásico que aparece alrededor de los 2 años exhibe un vínculo muy fuerte con el SNCB y acarrea un pronóstico más favorable que el meduloblastoma clásico esporádico.[85,86] Se diagnostica meduloblastoma en hasta 3 veces más hombres que mujeres con SNCB.[87] Así como con otras neoplasias malignas, el tratamiento del meduloblastoma con radiación ionizante produce múltiples CCB en el campo de radiación.[52,68] Otras neoplasias malignas notificadas son el carcinoma de ovario,[88] el fibrosarcoma ovárico,[89,90] el astrocitoma,[91] el melanoma,[92] la enfermedad de Hodgkin,[93,94] el rabdomiosarcoma[95] y el carcinoma nasosinusal indiferenciado.[96]
Los queratoquistes odontogénicos o tumores odontogénicos queratoquísticos (TOQ), según el nuevo nombre otorgado por el grupo de trabajo de la Organización Mundial de la Salud, son una de las características principales del SNCB.[97] El hallazgo de LOH clonal de genes supresores de tumores comunes, como PTCH1, respalda la transición en la terminología para reflejar un proceso neoplásico.[48] Menos de la mitad los TOQ en personas con SNCB exhiben LOH de PTCH1.[54,98] Los tumores están cubiertos por epitelio escamoso delgado y una capa corrugada delgada de paraqueratina. El aumento de la actividad mitótica en el epitelio tumoral y la posible gemación en la membrana basal con formación de quistes derivados de la pared tumoral quizás sea la causa de las tasas altas de recidiva después de la enucleación simple.[97,99] En una serie de casos reciente con 183 pacientes sometidos a extirpación de TOQ, en el 6 % se encontró asociación con SNCB.[97] En un estudio se analizó la tasa de variantes patogénicas de PTCH1 en los TOQ asociados al SNCB, y se encontró que 11 de 17 personas portaban variantes germinales patogénicas de PTCH1, y que otras 3 personas tenían variantes somáticas de este gen.[100] En las personas con variantes germinales patogénicas de PTCH1 los TOQ se presentaron a edad temprana. Entre el 65 % al 100 % de las personas con SNCB presentan TOQ,[56,101] y las tasas son más altas en mujeres jóvenes.[102]
Los hoyuelos palmoplantares son otro hallazgo importante en el CCB, y se presentan en el 70 % al 80 % de las personas con SNCB.[62] La presencia de estos hoyuelos en pacientes con CCB de inicio temprano y TOQ se considera diagnóstico de SNCB.[103]
Varios hallazgos radiológicos se relacionan con el SNCB, como la calcificación laminillar de la hoz cerebral;[104,105] las costillas fusionadas, muy separadas o bífidas;[106] y las radiolucencias con forma de llama o lesiones óseas pseudoquísticas en las falanges, el carpo, el tarso, los huesos largos, la pelvis y la bóveda craneal, que se encuentran en las imágenes radiológicas diagnósticas.[60] Las imágenes de las anomalías en las costillas quizás sean útiles cuando se estudia el diagnóstico de SNCB en niños pequeños, quienes quizás todavía no manifiesten por completo las características de este síndrome en el examen físico. La presencia de anormalidades esqueléticas afecta el número de CCB y la gravedad de estos en las personas afectadas. En un estudio, las personas con SNCB y manifestaciones esqueléticas presentaron una media de 120 más CCB que las personas con el síndrome pero sin manifestaciones esqueléticas. Las personas con 2 o más cambios esqueléticos también tuvieron un riesgo más alto de CCB avanzado o metastásico (OR, 2,45; IC 95 %, 1,01–5,91).[107]
En el Cuadro 2 se resume la frecuencia y la mediana de edad de inicio de las manifestaciones no malignas relacionadas con el SNCB.
| Hallazgo | Frecuencia (%) | Mediana de edad de inicio |
|---|---|---|
| Adaptación de un informe de Kimonis et al.[56]sobre 105 personas con SNCB atendidas en los Institutos Nacionales de la Salud entre 1985 y 1997. | ||
| Hoyuelos palmoplantares | 87 | Por lo general a los 10 años |
| Quistes queratogénicos en el maxilar | 74 | Por lo general a los 20 años |
| Puente en la silla turca | 68 | Congénito |
| Calcificación de la hoz cerebral | 65 | Por lo general a los 40 años |
| Macrocefalia | 50 | Congénito |
| Hipotelorismo | 42 | Congénito |
| Radiolucencias óseas en las manos | 30 | Congénito |
| Frente prominente | 27 | Congénito |
| Costillas bífidas | 26 | Congénito |
| Calcificación de la tienda del cerebelo | 20 | No notificado |
| Fibromas ováricos | 17 | 30 años |
| Hemivértebras | 15 | Congénito |
| Deformidad del tórax | 11 | Congénito |
| Fusión de cuerpos vertebrales | 10 | Congénito |
| Labio leporino y paladar hendido | 3 | Congénito |
Las personas con variantes patogénicas de PTCH2 a veces exhiben un fenotipo de SNCB menos intenso que las personas con variantes del gen PTCH1. Es posible que estas personas no presenten características como hoyuelos palmoplantares, macrocefalia, calcificación de la hoz cerebral, hipertelorismo y cara tosca.[108]
En 10 niños se describió un síndrome de microdeleción de 9p22.3 que afecta el locus PTCH1.[109] Todos los pacientes mostraban características faciales típicas del SNCB, como frente ancha; pero otras características eran variables, como craneosinostosis, hidrocefalia, macrosomía y retraso del desarrollo. En el momento del informe, ninguno tenía cáncer de piel de células basales. Dada la hemicigosidad del gen PTCH1, se presume que estos pacientes tienen un riesgo elevado de cáncer de piel de células basales.
Se han encontrado variantes germinales patogénicas de SUFU, un regulador inverso importante de la vía del erizo sónico (hedgehog) en un número pequeño de personas con un fenotipo clínico semejante al SNCB.[46,47,110] Estas variantes patogénicas se identificaron por primera vez en personas con meduloblastoma infantil,[111] la incidencia de meduloblastoma es mucho mayor en personas con SNCB asociado a variantes patogénicas de SUFU que en aquellas con variantes del gen PTCH1.[46] En un estudio se encontró que el 33 % de las personas con variantes patogénicas del gen SUFU que cumplían con los criterios clínicos de SNCB también tenían meduloblastomas.[61] En comparación, solo el 2,4 % de las personas con variantes patogénicas del gen PTCH1 tenían meduloblastomas (P = 0,009). Es posible que las variantes patogénicas de SUFU también se asocien con aumento de la predisposición al meningioma (variantes patogénicas de SUFU, 22 %; variantes patogénicas de PTCH1, 2 %).[61,70,110,112] En cambio, los queratoquistes odontogénicos mandibulares son menos frecuentes en esta población (variantes patogénicas de SUFU, 0 %; variantes patogénicas de PTCH1, 63 %). Algunos laboratorios clínicos ofrecen pruebas genéticas para las variantes patogénicas de SUFU en pacientes con SNCB. En una revisión de historias clínicas de niños con meduloblastomas portadores de variantes patogénicas de SUFU, se encontró que solo el 23 % (5 de 22) cumplió con los criterios SNCB.[113] El 36 % de estos niños murió al cabo de 1 año de recibir el diagnóstico de meduloblastoma, en su mayoría por progresión tumoral. Otros tumores subsecuentes también se notificaron en estos niños y en los familiares que también portaban variantes de SUFU, pero solo se notificó el caso de una persona con múltiples CCB. Este hallazgo indica que la presentación clínica del SNCB en portadores de variantes de SUFU es diferente de la que se observa en otras personas con variantes patogénicas de PTCH1. En este estudio, se encontraron variantes patogénicas de novo de SUFU en 3 de 22 niños.
Genes de reparación del DNA
Además de las variantes patogénicas de los genes asociados de manera primaria con el CCB, otros genes relacionados con el cáncer pueden conferir un aumento del riesgo de CCB. En un estudio de 61 personas con un número elevado de CCB (media, 11 CCB; intervalo, 6–65), se hizo una prueba genética para 29 genes de susceptibilidad al cáncer de penetrancia alta. Se encontraron 13 variantes patogénicas de 12 de las 61 personas (19,7 %). Este resultado fue más alto de lo esperado en comparación con las personas de la base de datos Exome Aggregation Consortium (ExAC) (3 %). Todos estos genes con variantes patogénicas repercutían en la reparación del DNA, lo que indica que los errores en las vías de reparación del DNA quizás aumentan el riesgo de CCB. De estas 61 personas, 21 (34,4 %) presentaban un antecedente de diagnóstico de otro tipo de cáncer, como melanoma, cáncer de mama, cáncer de colon y cáncer de próstata.
Xeroderma pigmentoso
El xeroderma pigmentoso (XP) es un trastorno hereditario de la reparación por escisión de nucleótidos que produce neoplasias malignas cutáneas en la primera década de la vida.[114] Las personas afectadas tienen aumento en la sensibilidad al sol, lo que se traduce en aumento del riesgo de CCE, CCB y melanomas. Para obtener más información, consultar la sección Xeroderma pigmentoso en la sección Carcinoma de células escamosas.
Síndromes raros
Síndrome de Rombo
El síndrome de Rombo es un trastorno genético o genodermatosis muy infrecuente que se vincula con el CCB. Se piensa que es un trastorno de herencia autosómica dominante y se han reportado 3 series de casos en la literatura médica.[115,116,117] Los exámenes cutáneos son normales hasta los 7 a 10 años, cuando se empieza a observar un eritema cianótico muy característico en los labios, las manos y los pies, además de atrofodermia vermiculada en las mejillas, con compromiso variable de los codos y el dorso de las manos y los pies.[115] El CCB aparece en la cuarta década.[115] Se ha descrito una textura granulosa de la piel característica, debido a la presencia de pápulas pequeñas amarillentas en la base de los folículos y atrofia folicular.[115,117] Otro hallazgo relacionado son las anormalidades en las pestañas y cejas, como ausencia, distribución irregular o desviación.[115,116] No se conoce la causa genética del síndrome de Rombo.
Síndrome de Bazex-Dupré-Christol
El síndrome de Bazex-Dupré-Christol es otra genodermatosis rara relacionada con el desarrollo de CCB, que cuenta con una documentación más completa que el síndrome de Rombo. La herencia exhibe un modelo dominante ligado al cromosoma X, y no se ha notificado transmisión de varón a varón.[118,119,120] La asignación regional del locus de interés al cromosoma Xq24-q27 se asocia con un puntaje de LOD de 5,26 en el locus DXS1192.[121] Investigaciones posteriores delimitaron la ubicación a un intervalo de 11.4-Mb en el cromosoma Xq25-27; sin embargo, el gen causal no se conoce.[122]
Las características físicas abarcan hipotricosis, hipohidrosis, milios, atrofodermia folicular en las mejillas y muchos CCB, que se manifiestan hacia el final de la segunda década de vida y el principio de la tercera.[118] Se han documentado cambios foliculares en el síndrome de Bazex-Dupré-Christol que abarcan disminución en la densidad del pelo en la cabeza y el cuerpo, melanización reducida,[123] apariencia retorcida o aplanada del tallo del pelo en el microscopio electrónico [124] y aumento del diámetro del tallo en el microscopio óptico polarizado.[120] Los milios, que son muy típicos durante la niñez, a veces desaparecen o disminuyen bastante en la pubertad.[120] Otras manifestaciones notificadas para este síndrome son los tricoepiteliomas, la hidradenitis supurativa, alas nasales hipoplásicas y columela prominente, el extremo de tejido blando del tabique nasal.[125,126]
Epidermólisis ampollosa simple
Un subtipo raro y grave de epidermólisis ampollosa simple (EBS), previamente conocido como Dowling-Meara (EBS-DM), se hereda principalmente de manera autosómica dominante y se asocia con variantes patogénicas de la queratina-5 (KRT5) o la queratina-14 (KRT14).[127,128] Este subtipo grave de EBS a veces causa la muerte durante la infancia temprana.[129] Su prevalencia estimada es de 0,02 casos por millón de personas en los Estados Unidos, y una incidencia de 1,16 casos por millón de nacidos vivos.[130] En un informe se citó una incidencia de CCB de 44 % a los 55 años en esta población.[131] Las personas que heredan 2 variantes patogénicas de EBS tal vez exhiban un fenotipo más grave.[132] Otros subtipos de EBS menos graves desde el punto de vista fenotípico también son causados por variantes patogénicas de KRT5 o KRT14.[127] Alrededor del 75 % de las personas con diagnóstico clínico de EBS (sin importar el subtipo) tienen variantes patogénicas de KRT5 o KRT14.[133]
Las características de los síndromes hereditarios relacionados con una predisposición al CCB se describen a continuación en el Cuadro 3.
| Síndrome | Herencia | Gen o locus cromosómico | Observaciones clínicas |
|---|---|---|---|
| AD = autosómica dominante; AR =autosómica recesivo; CCE = carcinoma de células escamosas; XD = dominante ligada al cromosoma X. | |||
| Síndrome de nevo de células basales, síndrome de Gorlin | AD | PTCH1,[134,135]PTCH2,[45]SUFU[70] | CCB (antes de los 20 años) |
| Síndrome de Rombo | AD | Desconocida | Milios, atrofodermia vermiculada, acrocianosis, tricoepiteliomas y CCB (edad de 30–40 años) |
| Síndrome de Bazex-Dupré-Christol | XD > AD | Xq24-27[121] | Hipotricosis (variable),[118]hipohidrosis, milios, atrofodermia folicular (dorso de las manos), y múltiples CCB (adolescentes y adultos jóvenes)[118] |
| Síndrome de Brooke-Spiegler | AD | CYLD[136,137] | Cilindroma (frente, cuero cabelludo, tronco y área púbica),[138,139]tricoepiteliomas (alrededor de la nariz), espiradenoma y CCB |
| CCB infundibuloquístico hereditario múltiple | AD[140] | Desconocida | Múltiples CCB (de tipo infundibuloquístico) |
| Síndrome de Schopf-Schultz-Passarge | AR > AD | Desconocida | Displasia ectodérmica (hipotricosis, hipodoncia ydistrofia ungueal[anoniquia y traquioniquia]), hidrocistomas de párpados, queratosis e hiperhidrosis palmoplantar y CBB[141] |
| Xeroderma pigmentoso | AR | XPA,XPB/ERCC3,XPC,XPD/ERCC2,XPE/DDB2,XPF/ERCC4,XPG/ERCC5 | CCE, CCB, melanoma, sensibilidad al sol intensa, anormalidades oftalmológicas y neurológicas |
| Variante de xeroderma pigmentoso | AR | POLH | CCE, CCB, melanoma, sensibilidad al sol intensa, anomalías oftalmológicas |
Para obtener más información, consultar la sección Síndrome de Brooke-Spiegler, tricoepitelioma múltiple familiar y cilindromatosis familiar.
Intervenciones
Detección
Como se detalla más adelante, el U.S. Preventive Services Task Force no recomienda la administración regular de exámenes de detección para identificar de manera temprana las neoplasias cutáneas malignas, incluso el CCB. Sin embargo, una vez que se detecta un CCB en la piel de una persona, la National Comprehensive cáncer Network recomienda que dicha persona se someta a exámenes cutáneos completos 2 veces al año o cada año durante los primeros 5 años posteriores al diagnóstico. Después de los primeros 5 años, se recomienda por lo menos un examen cutáneo cada año durante el resto de la vida.[142]
En el Cuadro 4 se resumen las guías de práctica clínica disponibles sobre la vigilancia en personas con SNCB.
| IRM = imágenes por resonancia magnética. | |
| Adaptación de Bree et al.[58]y Foulkes et al.[143] | |
| Para adultos: | |
| • IRM cerebral (inicial) | |
| • Exploración cutánea cada 4 meses | |
| • Radiografía panorámica del maxilar cada año | |
| • Evaluación neurológica (si hay antecedentes de meduloblastoma) | |
| • Ecografía pélvica (inicial) | |
| • Exploración ginecológica cada año | |
| • Evaluación nutricional | |
| • Evaluación fetal para hidrocefalia, macrocefalia o fibromas cardíacos durante el embarazo | |
| • Reducción al mínimo de la exposición a la radiación diagnóstica cuando sea posible | |
| Para menores de edad: | |
| • IRM cerebral (cada año hasta los 8 años)[58] | |
| • Riesgo bajo (PTCH1): no se hace un examen de detección radiográfico a menos que se encuentren hallazgos anormales en el examen neurológico, un cambio en la circunferencia de la cabeza y otros signos o síntomas inusuales[143] | |
| • Riesgo alto (SUFU): IRM cerebral cada 4 meses hasta los 3 años, luego cada 6 meses hasta los 5 años[143] | |
| • Ecocardiograma (inicial) | |
| • Exploración dermatológica (inicial) | |
| • Anual hasta los 10 años, aumento de la frecuencia una vez se diagnóstica el primer carcinoma de células basales[143] | |
| • Radiografía panorámica del maxilar (inicial y luego cada año cuando no se observan quistes; una vez se diagnostica el primer quiste se continúan cada 6 meses hasta los 21 años o hasta que no se encuentren quistes por 2 años consecutivos) | |
| • A partir de los 8 años de edad y después cada 12–18 meses[143] | |
| • Algunos dermatólogos recomiendan esperar hasta que se presenten síntomas para comenzar a obtener radiografías panorámicas con el fin de limitar la exposición a la radiación[143] | |
| • Radiografía de columna vertebral al año de edad o en el momento del diagnóstico (si hay anormalidades, se sigue el protocolo de escoliosis) | |
| • Ecografía pélvica en la menarquia o a los 18 años de edad | |
| • Evaluación auditiva, del habla y oftalmológica | |
| • Reducción al mínimo de la exposición a la radiación diagnóstica cuando sea posible | |
Nivel de evidencia: 5
Prevención primaria
Evitar la exposición solar excesiva, esporádica o acumulada es importante para reducir el riesgo de CCB y de otras neoplasias malignas cutáneas. Programar las actividades fuera de las horas pico de radiación UV, usar ropa y sombreros de protección solar y abundante protector solar, además de evitar a toda costa el uso de cámaras bronceadoras son pasos razonables para minimizar el riesgo futuro de cáncer de piel.[144] En los pacientes que tienen determinada susceptibilidad genética (como SNCB), evitar o reducir la exposición a radiación ionizante es esencial para reducir la carga tumoral en el futuro.
Nivel de evidencia: 2aii
Quimioprevención
Se ha explorado la función de varios retinoides sistémicos, como la isotretinoína y la acitretina, en la quimioprevención y el tratamiento de múltiples CCB, en especial en pacientes con SNCB. En un estudio del uso de isotretinoína en 12 pacientes con múltiples CCB, entre ellos 5 pacientes con SNCB, se observó regresión tumoral; la eficacia fue menor a medida que aumentó el diámetro del tumor.[145] Sin embargo, los resultados fueron insuficientes para recomendar el uso de retinoides sistémicos para el tratamiento del CCB. Se hizo seguimiento a otros 3 pacientes, incluso 1 con SNCB, para la evaluación a largo plazo de la quimioprevención con isotretinoína y se encontró reducción significativa en el número de tumores por año en el transcurso del tratamiento.[145] Aunque la tasa de formación de tumores tiende a incrementarse de manera abrupta una vez se interrumpe la terapia con retinoides sistémicos, en algunos pacientes la tasa sigue siendo más baja que la tasa anterior al tratamiento, lo que permite un abordaje y seguimiento más apropiados de las neoplasias malignas cutáneas.[145,146,147] En resumen, el uso de retinoides sistémicos para la quimioprevención del CCB es razonable en pacientes de riesgo alto, por ejemplo, aquellos con xeroderma pigmentoso. Para obtener más información, consultar la sección Carcinoma de células escamosas.
La carga tumoral acumulativa y en evolución se deben evaluar de manera cuidadosa por la posibilidad de una administración a largo plazo de un tipo de medicamento que tiene efectos secundarios acumulados e idiosincráticos. El perfil de efectos secundarios de los retinoides sistémicos hace que se recomiende la supervisión de un profesional con experiencia específica en la formulación de los retinoides sistémicos y que se sienta cómodo al formular este tipo de medicamentos. Sin embargo, en todas las mujeres en edad reproductiva, es esencial evitar de manera estricta el embarazo durante la administración del retinoide sistémico —hasta 1 mes después de interrumpir la isotretinoína y 3 años después de interrumpir la acitretina— para evitar malformaciones devastadoras y potencialmente mortales. En los Estados Unidos, la isotretinoína solo se puede recetar en el contexto del programa iPledge de la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA).
Nivel de evidencia (retinoides): 2aii
En un estudio de fase II en 41 pacientes con SNCB, se demostró que el vismodegib (inhibidor de la vía del erizo sónico hedgehog) reduce la tasa anual por paciente de formación de nuevos CCB que requieren cirugía.[148] El tratamiento con 150 mg de vismodegib oral al día también produjo regresión de los CCB que ya se habían formado. Si bien los pacientes tratados exhibieron una regresión visible de los tumores, se demostró la presencia de neoplasias malignas residuales microscópicas en el análisis de las biopsias del sitio tumoral, y progresión tumoral después de la interrupción del tratamiento. Los efectos adversos incluyeron alteraciones del gusto, calambres musculares, pérdida del cabello y adelgazamiento, que llevaron a la interrupción del medicamento en el 54 % de los participantes. En un estudio posterior de fase II y sin enmascaramiento participaron 37 pacientes de la misma cohorte que continuaron el vismodegib durante 36 meses.[149] Los pacientes tratados con vismodegib presentaron una media de incidencia más baja de CCE nuevos elegibles para cirugía en comparación con los pacientes que recibieron un placebo (P < 0,0001). Sin embargo, solo el 17 % de los pacientes toleraron el vismodegib continuo durante los 36 meses. Los tumores reaparecieron después de interrumpir el tratamiento, pero los pacientes que reanudaron el tratamiento presentaron respuesta tumoral. La duración del beneficio después de interrumpir el vismodegib fue proporcional a la duración y el cumplimiento de la administración del medicamento durante el tratamiento. El uso de esquemas posológicos intermitentes de vismodegib (8 semanas de administración y 8 semanas de descanso después de un periodo inicial de dosificación diaria de 24 semanas, o 12 semanas de administración y 8 semanas de descanso) es eficaz para la reducción de los CCB en la población con SNCB, aunque no se han establecido comparaciones entre la dosificación continua y los esquemas posológicos intermitentes.[150] A partir del perfil de efectos secundarios y la tasa de recidiva de la enfermedad luego de la interrupción del medicamento, se han puesto en marcha estudios sobre la dosificación ideal del vismodegib.
Nivel de evidencia (vismodegib): 1aii
En un ensayo clínico controlado con placebo y con enmascaramiento doble de fase III, se evaluaron los efectos de la nicotinamida oral (vitamina B3) en 386 personas con antecedentes de por lo menos 2 carcinomas de queratinocitos (CCB o CCE) en los 5 años previos al comienzo del estudio.[151] A los 12 meses de tratamiento, aquellos que tomaban nicotinamida 500 mg 2 veces al día exhibieron una reducción del 20 % en la incidencia de CCB nuevos (IC 95 %, 6–39 %; P = 0,12). La tasa de nuevos carcinomas de queratinocitos fue un 23 % más baja en el grupo de nicotinamida (IC 95 %, 4–38 %; P = 0,02) que en el grupo de placebo. No se observaron diferencias clínicas significativas en los efectos adversos entre los dos grupos, y no hubo evidencia de beneficio después de la interrupción del tratamiento con nicotinamida. Cabe destacar que este estudio no se llevó a cabo en una población con una predisposición genética al CCB identificada.
Nivel de evidencia (nicotinamida): 1aii
Tratamiento
El tratamiento de los CCB individuales en el SNCB por lo general es el mismo que para los cánceres de células basales esporádicos. Debido al gran número de lesiones en algunos pacientes, esto representa un reto quirúrgico. La terapia de campo con imiquimod o la terapia fotodinámica son opciones atractivas porque permiten el tratamiento de varios tumores al mismo tiempo.[152,153] Sin embargo, dada la radiosensibilidad de los pacientes al SNCB, se debe evitar la radioterapia como opción terapéutica para tumores grandes.[56] No se han llevado a cabo ensayos aleatorizados, pero se han publicado informes de casos donde se indica que la terapia de campo exhibe resultados parecidos al del cáncer de células basales esporádico, con tasas de éxito más altas para los cánceres superficiales que para los cánceres nodulares.[152,153]
Las directrices consensuadas para el uso de terapia fotodinámica con metilaminolevulinato en el SNCB recomiendan que mejor se reserve esta modalidad para el CCB superficial de todos los tamaños y para el CCB nodular de menos de 2 mm de grosor.[154] Es posible considerar el tratamiento mensual con terapia fotodinámica para estos pacientes según la indicación clínica.
Nivel de evidencia (imiquimod y terapia fotodinámica): 4
También se ha investigado el tratamiento tópico con LDE225, un agonista de Smoothened, para el tratamiento del CCB en un número pequeño de pacientes con SNCB y se notificaron resultados prometedores;[155] sin embargo, hasta la fecha, la FDA no ha autorizado el uso de este medicamento.
Nivel de evidencia (LDE225): 1
Además de los efectos en la prevención del CCB en pacientes con SNCB, es posible que el vismodegib también tenga un efecto paliativo en los TOQ que se encuentran en esta población. En un informe inicial se indicó que el uso de GDC-0449, un inhibidor de la vía del erizo sónico (hedgehog), conocido como vismodegib, produjo resolución de los TOQ en un paciente con SNCB.[156] En otro estudio pequeño se encontró que 4 de 6 pacientes que tomaron 150 mg de vismodegib al día exhibieron una reducción del tamaño de los TOQ.[157] Ninguno de los 6 pacientes del estudio presentaron TOQ nuevos o exhibieron aumento de tamaño de los TOQ durante el tratamiento, y 1 paciente presentó respuesta sostenida durante 9 meses después de la interrupción del tratamiento.
Nivel de evidencia (vismodegib): 3diii
Referencias:
- Miller DL, Weinstock MA: Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol 30 (5 Pt 1): 774-8, 1994.
- Gon A, Minelli L: Risk factors for basal cell carcinoma in a southern Brazilian population: a case-control study. Int J Dermatol 50 (10): 1286-90, 2011.
- Wu S, Han J, Li WQ, et al.: Basal-cell carcinoma incidence and associated risk factors in U.S. women and men. Am J Epidemiol 178 (6): 890-7, 2013.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Berlin NL, Cartmel B, Leffell DJ, et al.: Family history of skin cancer is associated with early-onset basal cell carcinoma independent of MC1R genotype. Cancer Epidemiol 39 (6): 1078-83, 2015.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Epstein E: Value of follow-up after treatment of basal cell carcinoma. Arch Dermatol 108 (6): 798-800, 1973.
- Møller R, Nielsen A, Reymann F: Multiple basal cell carcinoma and internal malignant tumors. Arch Dermatol 111 (5): 584-5, 1975.
- Bergstresser PR, Halprin KM: Multiple sequential skin cancers. The risk of skin cancer in patients with previous skin cancer. Arch Dermatol 111 (8): 995-6, 1975.
- Robinson JK: Risk of developing another basal cell carcinoma. A 5-year prospective study. Cancer 60 (1): 118-20, 1987.
- Greenberg ER, Baron JA, Stukel TA, et al.: A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med 323 (12): 789-95, 1990.
- Karagas MR, Stukel TA, Greenberg ER, et al.: Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. Skin Cancer Prevention Study Group. JAMA 267 (24): 3305-10, 1992.
- Cantwell MM, Murray LJ, Catney D, et al.: Second primary cancers in patients with skin cancer: a population-based study in Northern Ireland. Br J Cancer 100 (1): 174-7, 2009.
- Efird JT, Friedman GD, Habel L, et al.: Risk of subsequent cancer following invasive or in situ squamous cell skin cancer. Ann Epidemiol 12 (7): 469-75, 2002.
- Wheless L, Black J, Alberg AJ: Nonmelanoma skin cancer and the risk of second primary cancers: a systematic review. Cancer Epidemiol Biomarkers Prev 19 (7): 1686-95, 2010.
- Frisch M, Hjalgrim H, Olsen JH, et al.: Risk for subsequent cancer after diagnosis of basal-cell carcinoma. A population-based, epidemiologic study. Ann Intern Med 125 (10): 815-21, 1996.
- Cho HG, Kuo KY, Li S, et al.: Frequent basal cell cancer development is a clinical marker for inherited cancer susceptibility. JCI Insight 3 (15): , 2018.
- Small J, Wallace K, Hill EG, et al.: A cohort study of personal and family history of skin cancer in relation to future risk of non-cutaneous malignancies. Cancer Causes Control 30 (11): 1213-1221, 2019.
- Tuohimaa P, Pukkala E, Scélo G, et al.: Does solar exposure, as indicated by the non-melanoma skin cancers, protect from solid cancers: vitamin D as a possible explanation. Eur J Cancer 43 (11): 1701-12, 2007.
- de Vries E, Soerjomataram I, Houterman S, et al.: Decreased risk of prostate cancer after skin cancer diagnosis: a protective role of ultraviolet radiation? Am J Epidemiol 165 (8): 966-72, 2007.
- Grant WB: A meta-analysis of second cancers after a diagnosis of nonmelanoma skin cancer: additional evidence that solar ultraviolet-B irradiance reduces the risk of internal cancers. J Steroid Biochem Mol Biol 103 (3-5): 668-74, 2007.
- Soerjomataram I, Louwman WJ, Lemmens VE, et al.: Are patients with skin cancer at lower risk of developing colorectal or breast cancer? Am J Epidemiol 167 (12): 1421-9, 2008.
- Tabata T, Kornberg TB: Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76 (1): 89-102, 1994.
- Lum L, Beachy PA: The Hedgehog response network: sensors, switches, and routers. Science 304 (5678): 1755-9, 2004.
- Tojo M, Kiyosawa H, Iwatsuki K, et al.: Expression of the GLI2 oncogene and its isoforms in human basal cell carcinoma. Br J Dermatol 148 (5): 892-7, 2003.
- Gailani MR, Bale SJ, Leffell DJ, et al.: Developmental defects in Gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell 69 (1): 111-7, 1992.
- Shanley SM, Dawkins H, Wainwright BJ, et al.: Fine deletion mapping on the long arm of chromosome 9 in sporadic and familial basal cell carcinomas. Hum Mol Genet 4 (1): 129-33, 1995.
- Hahn H, Christiansen J, Wicking C, et al.: A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J Biol Chem 271 (21): 12125-8, 1996.
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, et al.: The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet 14 (1): 78-81, 1996.
- Wicking C, Shanley S, Smyth I, et al.: Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. Am J Hum Genet 60 (1): 21-6, 1997.
- Smyth I, Narang MA, Evans T, et al.: Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene in basal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet 8 (2): 291-7, 1999.
- Shakhova O, Leung C, van Montfort E, et al.: Lack of Rb and p53 delays cerebellar development and predisposes to large cell anaplastic medulloblastoma through amplification of N-Myc and Ptch2. Cancer Res 66 (10): 5190-200, 2006.
- Goodrich LV, Johnson RL, Milenkovic L, et al.: Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 10 (3): 301-12, 1996.
- Rahnama F, Toftgård R, Zaphiropoulos PG: Distinct roles of PTCH2 splice variants in Hedgehog signalling. Biochem J 378 (Pt 2): 325-34, 2004.
- Smith MJ, Evans DG: PTCH2 is not a strong candidate gene for gorlin syndrome predisposition. Fam Cancer 21 (3): 343-346, 2022.
- Wadt KA, Aoude LG, Johansson P, et al.: A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin Genet 88 (3): 267-72, 2015.
- Carbone M, Flores EG, Emi M, et al.: Combined Genetic and Genealogic Studies Uncover a Large BAP1 Cancer Syndrome Kindred Tracing Back Nine Generations to a Common Ancestor from the 1700s. PLoS Genet 11 (12): e1005633, 2015.
- de la Fouchardière A, Cabaret O, Savin L, et al.: Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet 88 (3): 273-7, 2015.
- Mochel MC, Piris A, Nose V, et al.: Loss of BAP1 Expression in Basal Cell Carcinomas in Patients With Germline BAP1 Mutations. Am J Clin Pathol 143 (6): 901-4, 2015.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Ferrucci LM, Cartmel B, Molinaro AM, et al.: Host phenotype characteristics and MC1R in relation to early-onset basal cell carcinoma. J Invest Dermatol 132 (4): 1272-9, 2012.
- Farndon PA, Del Mastro RG, Evans DG, et al.: Location of gene for Gorlin syndrome. Lancet 339 (8793): 581-2, 1992.
- Shimkets R, Gailani MR, Siu VM, et al.: Molecular analysis of chromosome 9q deletions in two Gorlin syndrome patients. Am J Hum Genet 59 (2): 417-22, 1996.
- Bale AE: Variable expressivity of patched mutations in flies and humans. Am J Hum Genet 60 (1): 10-2, 1997.
- Fan Z, Li J, Du J, et al.: A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet 45 (5): 303-8, 2008.
- Smith MJ, Beetz C, Williams SG, et al.: Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol 32 (36): 4155-61, 2014.
- Pastorino L, Ghiorzo P, Nasti S, et al.: Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 149A (7): 1539-43, 2009.
- Agaram NP, Collins BM, Barnes L, et al.: Molecular analysis to demonstrate that odontogenic keratocysts are neoplastic. Arch Pathol Lab Med 128 (3): 313-7, 2004.
- High A, Zedan W: Basal cell nevus syndrome. Curr Opin Oncol 17 (2): 160-6, 2005.
- Bacanli A, Ciftcioglu MA, Savas B, et al.: Nevoid basal cell carcinoma syndrome associated with unilateral renal agenesis: acceleration of basal cell carcinomas following radiotherapy. J Eur Acad Dermatol Venereol 19 (4): 510-1, 2005.
- Strong LC: Genetic and environmental interactions. Cancer 40 (4 Suppl): 1861-6, 1977.
- Evans DG, Birch JM, Orton CI: Brain tumours and the occurrence of severe invasive basal cell carcinoma in first degree relatives with Gorlin syndrome. Br J Neurosurg 5 (6): 643-6, 1991.
- Levanat S, Gorlin RJ, Fallet S, et al.: A two-hit model for developmental defects in Gorlin syndrome. Nat Genet 12 (1): 85-7, 1996.
- Pan S, Dong Q, Sun LS, et al.: Mechanisms of inactivation of PTCH1 gene in nevoid basal cell carcinoma syndrome: modification of the two-hit hypothesis. Clin Cancer Res 16 (2): 442-50, 2010.
- Evans DG, Ladusans EJ, Rimmer S, et al.: Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 30 (6): 460-4, 1993.
- Kimonis VE, Goldstein AM, Pastakia B, et al.: Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 69 (3): 299-308, 1997.
- Veenstra-Knol HE, Scheewe JH, van der Vlist GJ, et al.: Early recognition of basal cell naevus syndrome. Eur J Pediatr 164 (3): 126-30, 2005.
- Bree AF, Shah MR; BCNS Colloquium Group: Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A 155A (9): 2091-7, 2011.
- Klein RD, Dykas DJ, Bale AE: Clinical testing for the nevoid basal cell carcinoma syndrome in a DNA diagnostic laboratory. Genet Med 7 (9): 611-9, 2005 Nov-Dec.
- Kimonis VE, Mehta SG, Digiovanna JJ, et al.: Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med 6 (6): 495-502, 2004 Nov-Dec.
- Evans DG, Oudit D, Smith MJ, et al.: First evidence of genotype-phenotype correlations in Gorlin syndrome. J Med Genet 54 (8): 530-536, 2017.
- Shanley S, Ratcliffe J, Hockey A, et al.: Nevoid basal cell carcinoma syndrome: review of 118 affected individuals. Am J Med Genet 50 (3): 282-90, 1994.
- Scully RE, Galdabini JJ, McNeely BU: Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 14-1976. N Engl J Med 294 (14): 772-7, 1976.
- Ponti G, Pastorino L, Pollio A, et al.: Ameloblastoma: a neglected criterion for nevoid basal cell carcinoma (Gorlin) syndrome. Fam Cancer 11 (3): 411-8, 2012.
- Schwartz RA: Basal-cell-nevus syndrome and gastrointestinal polyposis. N Engl J Med 299 (1): 49, 1978.
- Totten JR: The multiple nevoid basal cell carcinoma syndrome. Report of its occurrence in four generations of a family. Cancer 46 (6): 1456-62, 1980.
- Jones KL, Wolf PL, Jensen P, et al.: The Gorlin syndrome: a genetically determined disorder associated with cardiac tumor. Am Heart J 111 (5): 1013-5, 1986.
- Gorlin RJ: Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 66 (2): 98-113, 1987.
- Mortimer PS, Geaney DP, Liddell K, et al.: Basal cell naevus syndrome and intracranial meningioma. J Neurol Neurosurg Psychiatry 47 (2): 210-2, 1984.
- Kijima C, Miyashita T, Suzuki M, et al.: Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer 11 (4): 565-70, 2012.
- Tamoney HJ: Basal cell nevoid syndrome. Am Surg 35 (4): 279-83, 1969.
- DiSanto S, Abt AB, Boal DK, et al.: Fetal rhabdomyoma and nevoid basal cell carcinoma syndrome. Pediatr Pathol 12 (3): 441-7, 1992 May-Jun.
- Korczak JF, Brahim JS, DiGiovanna JJ, et al.: Nevoid basal cell carcinoma syndrome with medulloblastoma in an African-American boy: a rare case illustrating gene-environment interaction. Am J Med Genet 69 (3): 309-14, 1997.
- Wolthers OD, Stellfeld M: Benign mesenchymoma in the trachea of a patient with the nevoid basal cell carcinoma syndrome. J Laryngol Otol 101 (5): 522-6, 1987.
- Ponti G, Manfredini M, Pastorino L, et al.: PTCH1 Germline Mutations and the Basaloid Follicular Hamartoma Values in the Tumor Spectrum of Basal Cell Carcinoma Syndrome (NBCCS). Anticancer Res 38 (1): 471-476, 2018.
- Iacono RP, Apuzzo ML, Davis RL, et al.: Multiple meningiomas following radiation therapy for medulloblastoma. Case report. J Neurosurg 55 (2): 282-6, 1981.
- Mack EE, Wilson CB: Meningiomas induced by high-dose cranial irradiation. J Neurosurg 79 (1): 28-31, 1993.
- Moss SD, Rockswold GL, Chou SN, et al.: Radiation-induced meningiomas in pediatric patients. Neurosurgery 22 (4): 758-61, 1988.
- Chiritescu E, Maloney ME: Acrochordons as a presenting sign of nevoid basal cell carcinoma syndrome. J Am Acad Dermatol 44 (5): 789-94, 2001.
- Tom WL, Hurley MY, Oliver DS, et al.: Features of basal cell carcinomas in basal cell nevus syndrome. Am J Med Genet A 155A (9): 2098-104, 2011.
- Lo Muzio L, Nocini PF, Savoia A, et al.: Nevoid basal cell carcinoma syndrome. Clinical findings in 37 Italian affected individuals. Clin Genet 55 (1): 34-40, 1999.
- Goldstein AM, Pastakia B, DiGiovanna JJ, et al.: Clinical findings in two African-American families with the nevoid basal cell carcinoma syndrome (NBCC). Am J Med Genet 50 (3): 272-81, 1994.
- Yasar B, Byers HJ, Smith MJ, et al.: Common variants modify the age of onset for basal cell carcinomas in Gorlin syndrome. Eur J Hum Genet 23 (5): 708-10, 2015.
- Mazzola CA, Pollack IF: Medulloblastoma. Curr Treat Options Neurol 5 (3): 189-198, 2003.
- Amlashi SF, Riffaud L, Brassier G, et al.: Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer 98 (3): 618-24, 2003.
- Cowan R, Hoban P, Kelsey A, et al.: The gene for the naevoid basal cell carcinoma syndrome acts as a tumour-suppressor gene in medulloblastoma. Br J Cancer 76 (2): 141-5, 1997.
- Evans DG, Farndon PA, Burnell LD, et al.: The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64 (5): 959-61, 1991.
- Berlin NI, Van Scott EJ, Clendenning WE, et al.: Basal cell nevus syndrome. Combined clinical staff conference at the National Institutes of Health. Ann Intern Med 64 (2): 403-21, 1966.
- Jackson R, Gardere S: Nevoid basal cell carcinoma syndrome. Can Med Assoc J 105 (8): 850 passim, 1971.
- Lindeberg H, Halaburt H, Larsen PO: The naevoid basal cell carcinoma syndrome. Clinical, biochemical and radiological aspects. J Maxillofac Surg 10 (4): 246-9, 1982.
- CAWSON RA, KERR GA: THE SYNDROME OF JAW CYSTS, BASAL CELL TUMOURS AND SKELETAL ABNORMALITIES. Proc R Soc Med 57: 799-801, 1964.
- Kedem A, Even-Paz Z, Freund M: Basal cell nevus syndrome associated with malignant melanoma of the iris. Dermatologica 140 (2): 99-106, 1970.
- Zvulunov A, Strother D, Zirbel G, et al.: Nevoid basal cell carcinoma syndrome. Report of a case with associated Hodgkin's disease. J Pediatr Hematol Oncol 17 (1): 66-70, 1995.
- Potaznik D, Steinherz P: Multiple nevoid basal cell carcinoma syndrome and Hodgkin's disease. Cancer 53 (12): 2713-5, 1984.
- Beddis IR, Mott MG, Bullimore J: Case report: nasopharyngeal rhabdomyosarcoma and Gorlin's naevoid basal cell carcinoma syndrome. Med Pediatr Oncol 11 (3): 178-9, 1983.
- Sobota A, Pena M, Santi M, et al.: Undifferentiated sinonasal carcinoma in a patient with nevoid basal cell carcinoma syndrome. Int J Surg Pathol 15 (3): 303-6, 2007.
- González-Alva P, Tanaka A, Oku Y, et al.: Keratocystic odontogenic tumor: a retrospective study of 183 cases. J Oral Sci 50 (2): 205-12, 2008.
- Suzuki M, Nagao K, Hatsuse H, et al.: Molecular pathogenesis of keratocystic odontogenic tumors developing in nevoid basal cell carcinoma syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol 116 (3): 348-53, 2013.
- Shear M: The aggressive nature of the odontogenic keratocyst: is it a benign cystic neoplasm? Part 1. Clinical and early experimental evidence of aggressive behaviour. Oral Oncol 38 (3): 219-26, 2002.
- Guo YY, Zhang JY, Li XF, et al.: PTCH1 gene mutations in Keratocystic odontogenic tumors: a study of 43 Chinese patients and a systematic review. PLoS One 8 (10): e77305, 2013.
- Gu XM, Zhao HS, Sun LS, et al.: PTCH mutations in sporadic and Gorlin-syndrome-related odontogenic keratocysts. J Dent Res 85 (9): 859-63, 2006.
- Lam KY, Chan AC: Odontogenic keratocysts: a clinicopathological study in Hong Kong Chinese. Laryngoscope 110 (8): 1328-32, 2000.
- North JP, McCalmont TH, LeBoit P: Palmar pits associated with the nevoid basal cell carcinoma syndrome. J Cutan Pathol 39 (8): 735-8, 2012.
- Chenevix-Trench G, Wicking C, Berkman J, et al.: Further localization of the gene for nevoid basal cell carcinoma syndrome (NBCCS) in 15 Australasian families: linkage and loss of heterozygosity. Am J Hum Genet 53 (3): 760-7, 1993.
- Ratcliffe JF, Shanley S, Ferguson J, et al.: The diagnostic implication of falcine calcification on plain skull radiographs of patients with basal cell naevus syndrome and the incidence of falcine calcification in their relatives and two control groups. Br J Radiol 68 (808): 361-8, 1995.
- Ratcliffe JF, Shanley S, Chenevix-Trench G: The prevalence of cervical and thoracic congenital skeletal abnormalities in basal cell naevus syndrome; a review of cervical and chest radiographs in 80 patients with BCNS. Br J Radiol 68 (810): 596-9, 1995.
- Betancourt NJ, Qian MF, Pickford JR, et al.: Gorlin Syndrome: Assessing Genotype-Phenotype Correlations and Analysis of Early Clinical Characteristics as Risk Factors for Disease Severity. J Clin Oncol 40 (19): 2119-2127, 2022.
- Fujii K, Ohashi H, Suzuki M, et al.: Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam Cancer 12 (4): 611-4, 2013.
- Muller EA, Aradhya S, Atkin JF, et al.: Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am J Med Genet A 158A (2): 391-9, 2012.
- Huq AJ, Walsh M, Rajagopalan B, et al.: Mutations in SUFU and PTCH1 genes may cause different cutaneous cancer predisposition syndromes: similar, but not the same. Fam Cancer 17 (4): 601-606, 2018.
- Brugières L, Remenieras A, Pierron G, et al.: High frequency of germline SUFU mutations in children with desmoplastic/nodular medulloblastoma younger than 3 years of age. J Clin Oncol 30 (17): 2087-93, 2012.
- Aavikko M, Li SP, Saarinen S, et al.: Loss of SUFU function in familial multiple meningioma. Am J Hum Genet 91 (3): 520-6, 2012.
- Guerrini-Rousseau L, Dufour C, Varlet P, et al.: Germline SUFU mutation carriers and medulloblastoma: clinical characteristics, cancer risk, and prognosis. Neuro Oncol 20 (8): 1122-1132, 2018.
- DiGiovanna JJ, Kraemer KH: Shining a light on xeroderma pigmentosum. J Invest Dermatol 132 (3 Pt 2): 785-96, 2012.
- Michaëlsson G, Olsson E, Westermark P: The Rombo syndrome: a familial disorder with vermiculate atrophoderma, milia, hypotrichosis, trichoepitheliomas, basal cell carcinomas and peripheral vasodilation with cyanosis. Acta Derm Venereol 61 (6): 497-503, 1981.
- van Steensel MA, Jaspers NG, Steijlen PM: A case of Rombo syndrome. Br J Dermatol 144 (6): 1215-8, 2001.
- Ashinoff R, Jacobson M, Belsito DV: Rombo syndrome: a second case report and review. J Am Acad Dermatol 28 (6): 1011-4, 1993.
- Viksnins P, Berlin A: Follicular atrophoderma and basal cell carcinomas: the Bazex syndrome. Arch Dermatol 113 (7): 948-51, 1977.
- Vabres P, de Prost Y: Bazex-Dupré-Christol syndrome: a possible diagnosis for basal cell carcinomas, coarse sparse hair, and milia. Am J Med Genet 45 (6): 786, 1993.
- Rapelanoro R, Taïeb A, Lacombe D: Congenital hypotrichosis and milia: report of a large family suggesting X-linked dominant inheritance. Am J Med Genet 52 (4): 487-90, 1994.
- Vabres P, Lacombe D, Rabinowitz LG, et al.: The gene for Bazex-Dupré-Christol syndrome maps to chromosome Xq. J Invest Dermatol 105 (1): 87-91, 1995.
- Parren LJ, Abuzahra F, Wagenvoort T, et al.: Linkage refinement of Bazex-Dupré-Christol syndrome to an 11·4-Mb interval on chromosome Xq25-27.1. Br J Dermatol 165 (1): 201-3, 2011.
- Parrish JA, Baden HP, Goldsmith LA, et al.: Studies of the density and the properties of the hair in a new inherited syndrome of hypotrichosis. Ann Hum Genet 35 (3): 349-56, 1972.
- Gould DJ, Barker DJ: Follicular atrophoderma with multiple basal cell carcinomas (Bazex). Br J Dermatol 99 (4): 431-5, 1978.
- Yung A, Newton-Bishop JA: A case of Bazex-Dupré-Christol syndrome associated with multiple genital trichoepitheliomas. Br J Dermatol 153 (3): 682-4, 2005.
- Kidd A, Carson L, Gregory DW, et al.: A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet 33 (6): 493-7, 1996.
- Arin MJ, Grimberg G, Schumann H, et al.: Identification of novel and known KRT5 and KRT14 mutations in 53 patients with epidermolysis bullosa simplex: correlation between genotype and phenotype. Br J Dermatol 162 (6): 1365-9, 2010.
- Has C, Bauer JW, Bodemer C, et al.: Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 183 (4): 614-627, 2020.
- Fine JD: Inherited epidermolysis bullosa. Orphanet J Rare Dis 5: 12, 2010.
- Fine JD: Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol 152 (11): 1231-1238, 2016.
- Fine JD, Johnson LB, Weiner M, et al.: Epidermolysis bullosa and the risk of life-threatening cancers: the National EB Registry experience, 1986-2006. J Am Acad Dermatol 60 (2): 203-11, 2009.
- García M, Santiago JL, Terrón A, et al.: Two novel recessive mutations in KRT14 identified in a cohort of 21 Spanish families with epidermolysis bullosa simplex. Br J Dermatol 165 (3): 683-92, 2011.
- Bolling MC, Lemmink HH, Jansen GH, et al.: Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol 164 (3): 637-44, 2011.
- Johnson RL, Rothman AL, Xie J, et al.: Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 272 (5268): 1668-71, 1996.
- Hahn H, Wicking C, Zaphiropoulous PG, et al.: Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85 (6): 841-51, 1996.
- Scheinfeld N, Hu G, Gill M, et al.: Identification of a recurrent mutation in the CYLD gene in Brooke-Spiegler syndrome. Clin Exp Dermatol 28 (5): 539-41, 2003.
- Bignell GR, Warren W, Seal S, et al.: Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 25 (2): 160-5, 2000.
- Weyers W, Nilles M, Eckert F, et al.: Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol 15 (2): 156-61, 1993.
- Rajan N, Langtry JA, Ashworth A, et al.: Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction. Arch Dermatol 145 (11): 1277-84, 2009.
- Requena L, Fariña MC, Robledo M, et al.: Multiple hereditary infundibulocystic basal cell carcinomas: a genodermatosis different from nevoid basal cell carcinoma syndrome. Arch Dermatol 135 (10): 1227-35, 1999.
- Nordin H, Månsson T, Svensson A: Familial occurrence of eccrine tumours in a family with ectodermal dysplasia. Acta Derm Venereol 68 (6): 523-30, 1988.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Basal Cell Skin Cancer. Version 2.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free subscription Last accessed June 24, 2024.
- Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.
- Lin JS, Eder M, Weinmann S, et al.: Behavioral Counseling to Prevent Skin Cancer: Systematic Evidence Review to Update the 2003 U.S. Preventive Services Task Force Recommendation. Agency for Healthcare Research and Quality, 2011. Report No.: 11-05152-EF-1. Also available online. Last accessed April 25, 2024.
- Peck GL, DiGiovanna JJ, Sarnoff DS, et al.: Treatment and prevention of basal cell carcinoma with oral isotretinoin. J Am Acad Dermatol 19 (1 Pt 2): 176-85, 1988.
- Goldberg LH, Hsu SH, Alcalay J: Effectiveness of isotretinoin in preventing the appearance of basal cell carcinomas in basal cell nevus syndrome. J Am Acad Dermatol 21 (1): 144-5, 1989.
- Cristofolini M, Zumiani G, Scappini P, et al.: Aromatic retinoid in the chemoprevention of the progression of nevoid basal-cell carcinoma syndrome. J Dermatol Surg Oncol 10 (10): 778-81, 1984.
- Tang JY, Mackay-Wiggan JM, Aszterbaum M, et al.: Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med 366 (23): 2180-8, 2012.
- Tang JY, Ally MS, Chanana AM, et al.: Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol 17 (12): 1720-1731, 2016.
- Dréno B, Kunstfeld R, Hauschild A, et al.: Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): a randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol 18 (3): 404-412, 2017.
- Chen AC, Martin AJ, Choy B, et al.: A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 373 (17): 1618-26, 2015.
- Stockfleth E, Ulrich C, Hauschild A, et al.: Successful treatment of basal cell carcinomas in a nevoid basal cell carcinoma syndrome with topical 5% imiquimod. Eur J Dermatol 12 (6): 569-72, 2002 Nov-Dec.
- Mougel F, Debarbieux S, Ronger-Savlé S, et al.: Methylaminolaevulinate photodynamic therapy in patients with multiple basal cell carcinomas in the setting of Gorlin-Goltz syndrome or after radiotherapy. Dermatology 219 (2): 138-42, 2009.
- Basset-Seguin N, Bissonnette R, Girard C, et al.: Consensus recommendations for the treatment of basal cell carcinomas in Gorlin syndrome with topical methylaminolaevulinate-photodynamic therapy. J Eur Acad Dermatol Venereol 28 (5): 626-32, 2014.
- Skvara H, Kalthoff F, Meingassner JG, et al.: Topical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol 131 (8): 1735-44, 2011.
- Goldberg LH, Landau JM, Moody MN, et al.: Resolution of odontogenic keratocysts of the jaw in basal cell nevus syndrome with GDC-0449. Arch Dermatol 147 (7): 839-41, 2011.
- Ally MS, Tang JY, Joseph T, et al.: The use of vismodegib to shrink keratocystic odontogenic tumors in patients with basal cell nevus syndrome. JAMA Dermatol 150 (5): 542-5, 2014.
Carcinoma de células escamosas
Introducción
El carcinoma de células escamosas (CCE) es el segundo tipo más común de cáncer de piel y representa cerca del 20 % de las neoplasias malignas cutáneas. Aunque la mayoría de los registros de cáncer no incluyen información sobre la incidencia de los carcinomas de queratinocitos (carcinoma de células basales [CCB] y CCE), la incidencia anual estimada oscila entre 1 millón a 5,4 millones de casos en los Estados Unidos.[1,2,3] En múltiples estudios se indica aumento del riesgo de CCE después del primer carcinoma de queratinocitos; en un metanálisis y revisión de 45 estudios se calculó que, después del diagnóstico de CCE primario, el 13,3 % de los pacientes presentará otro CCE (intervalo de confianza [IC] 95 %, 7,4 %–22,8 %).[4]
La muerte por este cáncer es infrecuente; sin embargo, la morbilidad y los costos del tratamiento son considerables.
Factores de riesgo del carcinoma de células escamosas
Exposición solar y otros factores de riesgo
La exposición al sol es el principal factor ambiental conocido que se relaciona con la formación del cáncer en todos los tipos de cáncer de piel; sin embargo, hay diferentes patrones de exposición al sol que se vinculan con cada tipo principal de cáncer de piel.[5,6,7] A diferencia del CCB, el CCE se vincula con exposición crónica a la radiación ultravioleta (UV), más que con la exposición intensa intermitente. La exposición ocupacional es el patrón característico de exposición solar que se vincula con el CCE.[5] Otros agentes y factores relacionados con el riesgo de CCE son las cámaras solares, el arsénico, la radiación terapéutica (como la terapia con psoraleno y radiación UVA para la psoriasis), la ulceración cutánea crónica y la inmunodepresión.[8,9,10,11,12,13,14,15,16] Para obtener más información sobre las exposiciones que pueden causar cáncer de piel en la población general, consultar Prevención del cáncer de piel.
Características de la piel
El CCB y el CCE, al igual que el melanoma, se presenta de manera más frecuente en personas de piel más clara que en personas de piel más oscura.[5,17] En un estudio de casos y controles con 415 casos y 415 controles se observaron hallazgos similares; al compararlas con las personas de tipo de piel Fitzpatrick I, aquellas con piel progresivamente más oscura presentaron reducción del riesgo de cáncer de piel (oportunidad relativa [OR], 0,6, 0,3 y 0,1, tipos de piel Fitzpatrick II, III y IV, respectivamente).[18] En el mismo estudio se observó que los ojos azules y el cabello rubio o rojo también se relacionaron con un aumento del riesgo de CCE, con OR cruda de 1,7 (IC 95 %, 1,2–2,3) para los ojos azules, 1,5 (IC 95 %, 1,1–2,1) para el cabello rubio y 2,2 (IC 95 %, 1,5–3,3) para el cabello rojo. Para obtener más información, consultar la sección Características pigmentarias en la sección Melanoma.
Sin embargo, el CCE también se presenta en personas de piel más oscura. En un registro asiático con sede en Singapur se notificó un aumento de cáncer de piel en esa zona geográfica, con una tasa de incidencia de 8,9 por 100 000 años-persona. Sin embargo, se observó que la incidencia de CCE estaba en descenso.[17] El CCE es la forma más común de cáncer de piel en personas negras en los Estados Unidos y en determinadas partes del África; la tasa de mortalidad de la enfermedad es relativamente alta en estas poblaciones.[19,20] Las características epidemiológicas y las estrategias de prevención del CCE en personas con piel más oscura todavía son objeto de investigación.
Las pecas en la piel y la reacción de la piel a la exposición solar son otros factores de riesgo identificados del CCE.[21] Las personas con muchas pecas en los antebrazos tenían un riesgo 4 veces más alto de CCE si las pecas moderadas o intensas estaban presentes desde la niñez o en la adultez.[22] El grado de riesgo del CCE se corresponde con el aumento en la cantidad de pecas. En este estudio, la incapacidad de la piel para broncearse y la propensión a quemarse también se relacionaron de manera significativa con el riesgo de CCE (OR de 1,54 para quemaduras o ampollas y de 12,44 para pecas o incapacidad de bronceado).
La presencia de cicatrices en la piel también aumenta el riesgo de CCE, aunque la carcinogénesis en este entorno quizás tome años o incluso decenios. Los CCE que se presentan en heridas crónicas se conocen como úlceras de Marjolin. La media de tiempo hasta la formación del carcinoma en estas heridas es de 26 años.[23] En un informe de casos se notificó la presencia de cáncer en una herida de 59 años atrás.[24]
Inmunodepresión
La inmunodepresión también contribuye a la formación de CCB y CCE. En las personas receptores de trasplantes de órganos sólidos, el riesgo de CCE es de 65 a 250 veces más alto, y el riesgo de CCB es 10 veces más alto que el observado en la población general, aunque el riesgo varía según el tipo de trasplante y el fármaco inmunodepresor.[25,26,27,28] Los CCB y CCE en pacientes de riesgo alto (receptores de trasplantes de órganos y pacientes con leucemia linfocítica crónica) se presentan a temprana edad, son más frecuentes y de mayor malignidad, además de que exhiben un riesgo más alto de recidiva y diseminación metastásica que los cánceres de la población general.[29,30] Además, hay un riesgo más alto de CCE secundarios.[31,32] En un estudio, más del 65 % de los receptores de trasplantes de riñón presentaron otros CCE después del diagnóstico inicial.[31] Entre los pacientes de Medicare con un sistema inmunitario intacto, los CCB se presentan de manera tan frecuente como los CCE;[3] en receptores de trasplantes, los CCE sobrepasan en cantidad a los CCB en una proporción 2:1.
Antecedentes personales de cáncer de piel de células basales, de células escamosas y melanoma
Los antecedentes personales de CCB o CCE se relacionan de manera importante con un CCB o CCE posterior. En un estudio en Irlanda se observó que las personas con antecedentes de CCB tenían una incidencia 14 % más alta de CCE subsiguiente; en los hombres con antecedentes de CCB, el riesgo de CCE subsiguiente fue un 27 % más alto.[33] En el mismo informe, las personas con melanoma también fueron 2,5 veces más propensas a notificar un CCE posterior. Hay un aumento aproximado del 20 % en el riesgo de una lesión subsiguiente en el primer año posterior al diagnóstico de un cáncer de piel. La media de edad de aparición de estos cánceres se sitúa en la mitad de la sexta década de vida.[34,35,36,37,38,39]
En un estudio Suizo de 224 probandos con melanoma y 944 familiares de primer grado (FPG) de 154 familias con CDKN2A normal y 11 680 controles emparejados, se observó que los antecedentes personales y familiares de melanoma aumentan el riesgo de SCC, con un riesgo relativo (RR) de 9,1 (IC 95 %, 6,0–13,7) para los antecedentes personales y de 3,4 (IC 95 %, 2,2–5,2) para los antecedentes familiares.[40] En otro estudio de 216 115 personas del Nurses' Health Study, el Nurses' Health Study 2 y el Health Professionals Follow-Up Study, se encontró que las personas con antecedentes familiares de melanoma exhibieron un 22 % de aumento del riesgo de CCE (cociente de riesgos instantáneos, 1,22; IC 95 %, 1,06–1,40).[41]
Historia familiar de carcinoma de células escamosas o lesiones premalignas asociadas
Si bien la bibliografía es escasa sobre este tema, una historia familiar de CCE aumenta el riesgo de CCE en los FPG. En un estudio independiente de encuestas se analizaron 415 casos de CCE y 415 controles, el riesgo de CCE aumentó en las personas con una historia familiar de CCE (OR ajustada, 3,4; IC 95 %, 1,0–11,6), incluso después del ajuste por tipo de piel, color de cabello y color de ojos.[18] El riesgo aumentó hasta una OR de 5,6 en quienes tenían una historia familiar de melanoma (IC 95 %, 1,6–19,7), 9,8 en quienes tenían una historia familiar de CCB (IC 95 %, 2,6–36,8), y 10,5 en quienes tenían una historia familiar de varios tipos de cáncer de piel (IC 95 %, 2,7–29,6). En la revisión de la Swedish Family Center Database se observó que las personas con al menos un hermano, hermana o progenitor afectados por CCE, CCE in situ (enfermedad de Bowen) o queratosis actínica presentaban un aumento doble o triple del riesgo de CCE invasivo e in situ que la población general.[42,43] Un número alto de tumores en los progenitores se relacionó con aumento del riesgo en la descendencia. Cabe destacar que el diagnóstico del probando a una edad más temprana no se relacionó de manera sistemática con una tendencia de aumento en la incidencia de CCE en los FPG, como se esperaría en la mayoría de los síndromes hereditarios debido a las variantes patogénicas germinales. En otros análisis de la base de datos de la población sueca se calculó que el efecto del riesgo genético es del 8 % y el efecto del entorno familiar es del 18 %.[44] Es decir, que el entorno ambiental y los factores del comportamiento explican al menos en parte el agrupamiento familiar del CCE.
En un estudio sobre la heredabilidad del cáncer en 80 309 gemelos monocigóticos y 123 382 gemelos dicigóticos, se observó que los NMSC tienen una heredabilidad del 43 % (IC 95 %, 26–59 %), lo que indica que casi la mitad del riesgo de este cáncer se debe a factores hereditarios.[45] Además, el riesgo acumulado del NMSC fue 1,9 veces más alto para los gemelos monocigóticos que para los gemelos dicigóticos (IC 95 %, 1,8–2,0).[45]
Síndromes y genes asociados con la predisposición al carcinoma de células escamosas
En otras partes de este resumen se definen los principales genes que son necesarios y suficientes para que aparezca la enfermedad, además de las variantes patogénicas de estos genes que se consideran causales de enfermedad. Los trastornos producidos por las variantes patogénicas de un solo gen en determinadas familias conllevan un riesgo muy alto de enfermedad y son relativamente infrecuentes. La influencia del medio ambiente en la aparición de la enfermedad en personas con estos trastornos de un solo gen a menudo es muy difícil de determinar debido a la rareza de la variante genética.
La identificación de un factor de riesgo ambiental fuerte, como la exposición crónica a la radiación UV, dificulta el análisis de la causalidad genética del CCE cutáneo. Aunque el riesgo que se deriva de la exposición a la radiación UV es bien conocido, la cuantificación del riesgo atribuible para la formación del cáncer es un reto. Además, la determinación de los casos de CCE de piel no siempre es sencilla. En muchos registros y estudios epidemiológicos no se evalúa por completo la incidencia del CCE de la piel debido a los siguientes aspectos: 1) es una práctica común tratar lesiones sospechosas de CCE incluso cuando no se cuenta con diagnóstico por biopsia y 2) el potencial de metástasis es relativamente bajo. Además, el NMSC se suele excluir de manera rutinaria de los principales registros del cáncer, como el Surveillance, Epidemiology, and End Results.
Teniendo en cuenta estas consideraciones, el análisis que sigue se enfoca en los genes relacionados con trastornos que aumentan la incidencia de cáncer de piel.
Las características de los principales síndromes hereditarios relacionados con una predisposición al CCE se describen en el Cuadro 5.
| Afección | Genes | Vías |
|---|---|---|
| SWI/SNF =SWItch/Sucrose Non-Fermentable. | ||
| a Información de Loh et al.[46] | ||
| Síndrome de Bloom | BLM/RECQL3 | Estabilidad cromosómica |
| Síndrome de Chediak-Higashi | LYST | Regulación del transporte lisosómico |
| Disqueratosis congénita | DKC1,TERC,TINF2,NHP2/NOLA2,NOP10/NOLA3,TERT,WRAP53,C16orf57,RTEL1 | Mantenimiento y transporte detelómeros |
| Epidermólisis ampollosa distrófica(subtipos de herenciaautosómica dominanteyautosómica recesiva) | COL7A1 | Anclaje de colágeno de la membrana basal a la dermis |
| Enfermedad de Elejalde | MYO5A | Transporte de gránulos de pigmentos |
| Epidermodisplasia verruciforme | EVER1/TMC6,EVER2/TMC8 | Transducción de señales en el retículo endoplasmático |
| Anemia de Fanconi | FANCA,FANCB,FANCC,FANCD1/BRCA2,FANCD2,FANCE,FANCF,FANCG/XRCC9,FANCI,FANCJ/BRIP1/BACH1,FANCL,FANCM,FANCN/PALB2,FANCO/RAD51C,FANCP/SLX4/BTBD12,FANCQ/ERCC4/XPF,FANCS/BRCA1 | Reparación delDNA |
| Síndrome de Griscelli(tipo 1, tipo 2 y tipo 3) | MYO5A,RAB27A,MLPH | Transporte de gránulos de pigmentos |
| Síndrome de Hermansky-Pudlak | HPS1,HPS2/AP3B1,HPS3,HPS4,HPS5,HPS6,HPS7/DTNBP1,HPS8/BLOC1S3,HPS9/BLOC1S6,HPS10/AP3D1 | Almacenamiento de melanosomas y lisosomas |
| Síndrome de Hurieza | SMARCAD1 | vía SWI/SNF reguladora de cromatina |
| Epidermólisis ampollosa de la unión | LAMA3,LAMB3,LAMC2,COL17A1 | Tejido conjuntivo |
| Epitelioma escamoso autocurativo múltiple (síndrome de Ferguson-Smith) | TGFBR1 | Señalización de factores de crecimiento |
| Albinismo oculocutáneo(tipo IA, tipo IB, tipo II, tipo III, tipo IV, tipo V, tipo VI y tipo VII) | TYR,OCA2,TYRP1,SLC45A2/MATP/OCA4, locus 4q24,SLC24A5,C10Orf11 | Síntesis de melanina |
| Síndrome de Rothmund-Thomson de tipo 1 | ANAPC1 | Ciclo celular |
| Síndrome de Rothmund-Thomson de tipo 2 | RECQL4,C16orf57 | Estabilidad cromosómica |
| Síndrome de Werner | WRN/RECQL2 | Estabilidad cromosómica |
| Xeroderma pigmentoso(grupos de complementación A, B, C, D, E, F y G) | XPA,XPB/ERCC3,XPC,XPD/ERCC2,XPE/DDB2,XPF/ERCC4,XPG/ERCC5 | Reparación de la escisión de nucleótidos |
| Variante de xeroderma pigmentoso | POLH | Polimerasa propensa a error |
Xeroderma pigmentoso
El xeroderma pigmentoso (XP) es un trastorno hereditario de la reparación por escisión de nucleótidos que produce neoplasias malignas cutáneas en la primera década de la vida.[47] Las personas afectadas tienen aumento de la sensibilidad al sol, lo que se traduce en aumento del riesgo de CCE, CCB y melanomas. En un informe se encontró que los carcinomas de queratinocitos (CCB y CCE) aumentaron 150 veces en personas con XP; en menores de 20 años, la prevalencia estimada es de 5000 a 10 000 veces el valor esperado en la población general.[48,49]
La historia natural de esta enfermedad comienza en el primer año de vida, cuando la sensibilidad al sol se hace evidente, y aparece xerosis (piel seca) y cambios pigmentarios en la piel expuesta al sol. Cerca de la mitad de los pacientes de XP tienen antecedentes de quemaduras graves con exposición solar mínima. Otros pacientes con XP no tienen esta reacción, pero presentan pigmentación en forma de pecas antes de los 2 años en sitios expuestos al sol. Estas manifestaciones progresan a atrofia cutánea y formación de telangiectasias. Cerca de la mitad de las personas con este trastorno presentarán CCB o CCE, y alrededor de un cuarto de estas personas padecerá de melanoma.[48] Cuando no se evita la exposición al sol, la mediana de edad de diagnóstico de cualquier cáncer de piel es de 8 a 9 años.[48,49,50] En promedio, los CCB y los CCE se presentan a una edad más temprana que el melanoma en la población con XP.[49]
Las manifestaciones no cutáneas de XP abarcan anomalías oftalmológicas, neurológicas y del envejecimiento. Las anomalías de la córnea y los párpados que se presentan en la XP se vinculan con exposición a la radiación UV. Los ejemplos de anomalías oftalmológicas incluyen queratitis, opacidad corneal, ectropión, entropión, hiperpigmentación de los párpados, pérdida de las pestañas y cáncer (incluso cáncer de conjuntiva y córnea).[51] Alrededor del 25 % de los pacientes con XP evaluados en los Institutos Nacionales de la Salud (NIH) entre 1971 y 2009 exhibieron degeneración neurológica progresiva.[49] Las características de la degeneración neurológica incluyen microcefalia, hipoacusia neurosensorial progresiva, disminución de los reflejos tendinosos profundos, convulsiones y alteraciones cognitivas. La degeneración neurológica, que se observa con mayor frecuencia en personas con grupos de complementación XPA y XPD, se vincula con una expectativa de vida más corta (mediana de edad de muerte de 29 años en personas con degeneración neurológica y de 37 años en personas sin degeneración neurológica).[49] Un subgrupo de pacientes con manifestaciones cutáneas y oculares de la XP además presentan alteraciones neurológicas graves, enanismo y retraso del desarrollo. Esta forma grave de XP se conoce como síndrome de De Sanctis-Cacchione. Las personas con XP a veces exhiben diferencias en el envejecimiento. En un estudio pequeño, 18 mujeres posmenopáusicas con XP exhibieron envejecimiento prematuro, con una mediana de edad de menopausia de 29,5 años (intervalo, 18–49,5 años) en el grupo de XP en comparación con 52,9 años en la población general de los Estados Unidos.[52]
En personas con XP también se han notificado varias neoplasias no cutáneas, en su mayoría CCE de la punta de la lengua, cáncer del sistema nervioso central, cánceres hematológicos, cáncer de tiroides, cánceres ginecológicos y cáncer de pulmón en personas que fuman.[48,53,54] Se estima que el RR de estos tipos de cáncer es 34 a 50 veces más alto en personas con XP que en la población general.[48,54] En un informe de 434 pacientes con XP de 4 cohortes internacionales (Estados Unidos, Reino Unido, Francia y Brasil) se encontró una tasa interna de cáncer del 11,3 % en personas con XP, con un promedio de edad de diagnóstico 50 años menor que en las personas de la población general de los Estados Unidos. Los pacientes con variantes patogénicas del gen XPC (en especial, aquellas de África del Norte que tienen la variante patogénica con efecto fundador delTG) presentaron un riesgo de cáncer más elevado que quienes tenían variantes patogénicas de otros genes de la XP.[54]
La herencia de XP es autosómica recesiva. Se han identificado 7 grupos de complementación asociados con este trastorno. Cerca del 40 % de los casos de XP observados en los NIH fueron de XPC. Se observaron variantes patogénicas de ERCC2 (XPD) en casi el 20 %. El grupo de complementación A, debido a una variante patogénica del gen XPA, representa cerca del 10 % de los casos.[49] Otros genes en este trastorno son ERCC3 (XPB), DDB2 (XPE), ERCC4 (XPF) y ERCC5 (XPG). Se describió un grupo XPH, pero ahora se considera que es un subgrupo del grupo XPD.[55] Las personas heterocigotas para variantes patogénicas de los genes de XP por lo general son asintomáticas.[56] Sin embargo, en un estudio se notificó un aumento triple en el CCB en personas japonesas heterocigotas para variantes patogénicas de XPA.[57] En poblaciones de África del norte, se han encontrado variantes patogénicas con efecto fundador de XPA (R228A) y XPC (V548A fs X572), y en población de África del Este (Mahori) se encontró una variante patogénica con efecto fundador de XPC que produce una alteración en el sitio de empalme (IVS 12-1G>C). Se ha planteado que sería apropiado usar un examen de detección específico de dichas variantes patogénicas en estas poblaciones.[58,59,60,61] Una variante patogénica con efecto fundador en el sitio aceptador del empalme 3' del intrón 3 en el gen XPA está presente en alrededor del 1 % de la población de Japón, lo que equivale a casi 1 millón de personas.[62]
La función de los genes XP es reconocer y reparar los fotoproductos de la radiación UV. Los principales fotoproductos se forman en las pirimidinas adyacentes y constan de dímeros de ciclobutano y fotoproductos pirimidina-pirimidona (6-4). El producto de XPC participa en la identificación inicial del daño al DNA; se une a la lesión y actúa como un marcador para la reparación posterior. La proteína DDB2 (XPE) también participa en este proceso y funciona con XPC. El producto del gen XPA mantiene regiones de una sola hebra durante la reparación y actúa con el complejo del factor transcripción TFIII. El complejo TFIIH incluye los productos génicos de ERCC3 (XPB) y ERCC2 (XPD), que funcionan como helicasas de DNA durante el desenrollado del DNA. Las proteínas ERCC4 (XPF) y ERCC5 (XPG) actúan como endonucleasas del DNA para crear muescas en una sola cadena en los extremos 5' y 3' del DNA dañado, lo que conlleva la escisión de 28 a 30 nucleótidos, que incluye el fotoproducto. Las polimerasas de DNA reemplazan la lesión con la secuencia correcta y una ligasa de DNA completa la reparación.[47,63]
Una variante de XP que se asocia con variantes patogénicas de POLH (XPV) es responsable de alrededor del 10 % de los casos reportados.[64] Este gen codifica la polimerasa de derivación propensa a error (polimerasa eta) que, contrario a los otros genes asociados con XP, no participa en la reparación de la escisión de nucleótidos. Las personas con variantes patogénicas de la polimerasa eta presentan los mismos hallazgos cutáneos y oculares que otros pacientes con XP, pero no exhiben degeneración neurológica progresiva.[65] Se encontró una variante patogénica con efecto fundador que produce una deleción del exón 10 en 16 de 16 personas de 10 familias consanguíneas de Túnez.[66]
El trabajo de investigación sobre las correlaciones entre genotipo y fenotipo en los grupos de complementación de XP continúa; sin embargo, la evidencia indica que la variante patogénica específica quizás tenga más influencia sobre el fenotipo que el grupo de complementación.[47,67] Las características diferenciadoras principales son la presencia o ausencia de quemaduras en respuesta a exposición solar mínima, el cáncer de piel y las anomalías neurológicas progresivas. Todos los grupos de complementación se caracterizan por la presencia de neoplasias cutáneas, pero los cánceres de piel son más comunes en los grupos XPC, XPE y XPV.[67]. Hay variación clínica adicional dentro de cada grupo de complementación. Se ha descrito una alteración neurológica leve a grave en personas con variantes patogénicas de XPA. Las personas con variantes patogénicas de XPA en la región de unión al DNA (aminoácidos 98–219) a veces exhiben una presentación clínica más grave que incluye manifestaciones neurológicas.[68] Las personas en el grupo de complementación XPC tienen incidencias más elevadas de lesión ocular.[67] Un número muy pequeño de personas en los grupos de complementación XPB, XPD y XPG presentan un complejo de xeroderma pigmentoso con síndrome de Cockayne (XP-CS). Estas personas tienen características de ambos trastornos, como una mayor predisposición a las neoplasias cutáneas y retraso del desarrollo, deterioro visual y auditivo, y disfunción del sistema nervioso central y periférico. Cabe señalar que las personas con síndrome de Cockayne sin XP no exhiben aumento del riesgo de cáncer.[69] De manera parecida, la tricotiodistrofia (TTD) es otro trastorno genético que ocurre en combinación con XP. Las personas afectadas únicamente por TTD no exhiben aumento en la incidencia de cáncer, pero algunas personas con XP/TTD exhiben un aumento del riesgo de neoplasias cutáneas. Los grupos de complementación vinculados con XP/TTD (XPD y XPB) y XP-CS (XPB, XPD y XPG) se relacionan con defectos en la reparación de escisión de nucleótidos unida a la transcripción y en la reparación de escisión de nucleótidos de todo el genoma. En contraste, los grupos de complementación C y E de XP solo presentan defectos en la reparación de la escisión de nucleótidos de todo el genoma.[47,70] Además, las personas con los grupos de complementación XPA, XPD y XPG quizás exhiban anormalidades neurológicas graves sin síntomas del síndrome de Cockayne ni TTD. El síndrome cerebro-oculo-facio-esquelético, que se ha descrito con unas variantes patogénicas de ERCC2 (XPD) o XP-CS, no parece conferir un aumento del riesgo de cáncer de piel.[71,72,73,74]
El diagnóstico de XP se establece a partir de los hallazgos clínicos y la historia familiar. Se han desarrollado ensayos funcionales para evaluar la capacidad de reparación del DNA después de la exposición a la radiación, pero estas pruebas no están disponibles en el ámbito clínico en los Estados Unidos. El uso de pruebas genéticas clínicas con análisis de secuencia para identificar variantes patogénicas está disponible para múltiples genes asociados con XP; la lista se encuentra en el NIH Genetic Testing Registry.
Epitelioma escamoso autocurativo múltiple (síndrome de Ferguson-Smith)
El epitelioma escamoso autocurativo múltiple (MSSE) o síndrome de Ferguson-Smith, descrito por primera vez en 1934, se caracteriza por tumores de piel invasores que son histológicamente idénticos al CCE cutáneo esporádico, pero que resuelven de manera espontánea sin intervención. El análisis de ligamiento de familias afectadas exhibe una asociación con el brazo largo del cromosoma 9, y el análisis del haplotipo ubica el gen 9q22.3 entre D9S197 y D9S1809.[75] El receptor beta del factor de crecimiento transformante 1 (TGFBR1) se identificó mediante secuenciación de última generación como el gen responsable del MSSE. Las variantes patogénicas de pérdida de función del gen TGFBR1 se identificaron en 18 de 22 familias afectadas.[76] Se han identificado variantes de ganancia de función de TGFBR1 relacionadas con síndromes similares a Marfan, como el síndrome de Loeys-Dietz, para el que no se ha descrito aumento en el riesgo de cáncer de piel.
En esta ubicación genómica se encontró pérdida de heterocigosidad somática en el CCE relacionado con el síndrome de Ferguson-Smith, lo que indica que TGFBR1 puede actuar como gen supresor de tumores.[77] El brazo largo del cromosoma 9 también ha demostrado ser un sitio de interés en el CCE esporádico. Se encontró que hasta el 65 % de los CCE esporádicos exhiben pérdida de heterocigosidad en 9q22.3 entre D9S162 y D9S165.[77]
Albinismo oculocutáneo
El albinismo es un factor de riesgo importante para el cáncer de piel en personas de ascendencia africana.[20,78] En un informe se describió una cohorte de 350 personas con albinismo en Tanzania donde se identificaron 104 casos de cáncer cutáneo; entre ellos, 100 CCE, 3 CCB y 1 melanoma.[79] La mediana de edad para esta población fue de 10 años. Se observaron proporciones similares de diagnóstico de cáncer de piel en una población nigeriana, con un 62 % de las neoplasias malignas dermatológicas diagnosticadas como CCE, un 16 % como melanoma y un 8 % como CCB.[20] Cabe señalar que se encontraron algunos melanomas sin melanina en personas con albinismo.[80]
El CCE que se presenta a edades muy tempranas es una característica distintiva del albinismo oculocutáneo. En una cohorte de casi 1000 pacientes nigerianos con albinismo, todos presentaban lesiones cutáneas malignas o premalignas a los 20 años de edad.[81]
Se sabe que dos tipos de albinismo oculocutáneo se relacionan con un aumento del riesgo de CCE en la piel. El albinismo oculocutáneo de tipo 1, o albinismo relacionado con la tirosinasa, se produce por variantes patogénicas del gen de la tirosinasa TYR, ubicado en el brazo largo del cromosoma 11. Este tipo de albinismo explica alrededor de la mitad de los casos en personas de ascendencia europea.[82] El gen OCA2, que también se conoce como gen P, se encuentra alterado en el albinismo oculocutáneo de tipo 2 o albinismo positivo para tirosinasa. Ambos trastornos son autosómico recesivos, con heterocigosis compuesta frecuente. Una deleción intragénica de 2,7 kb en el gen OCA2 establece una variante patogénica con efecto fundador común en África subsahariana, que explica la mayoría de los casos de albinismo relacionado con OCA2 en esta región.[83]
La tirosinasa actúa como una enzima esencial para la síntesis de melanina en los melanocitos. Una variante de este gen propia del albinismo oculocutáneo de tipo 1 produce proteínas con poca o nula actividad, que corresponden a los fenotipos OCA1B y OCA1A, respectivamente. Las personas con OCA1B nacen con un color claro en la piel, el cabello y los ojos, y luego adquieren un poco de pigmento durante la vida; por el contrario, las personas con OCA1A no adquieren pigmentación con la edad.
El producto del gen OCA2 es una proteína que se encuentra en la membrana de los melanosomas. Aunque se desconoce su función específica, participa en el mantenimiento de la estructura o el pH de este entorno.[84] Los modelos murinos con variantes de este gen presentan disminución significativa en la producción de melanina en comparación con los controles normales.[85] En un estudio internacional de personas con albinismo, se encontraron variantes bialélicas de OCA2 en el 17 % de los participantes.[86]
Las variantes genéticas de SLC45A2 (MATP asociada a OCA4), SLC24A5 (asociada a OCA6) y TYRP1 (proteína 1 relacionada con la tirosinasa asociada a OCA3) se vinculan con tipos menos comunes de albinismo oculocutáneo. La incidencia reportada de alteraciones en estos genes en una población internacional de pacientes con albinismo fue del 7 % para SLC45A2, 1 % para TYRP1, y menos de 0,5 % para SLC24A5.[86] El gen SLC45A2 se encuentra en el 24 % de los casos de albinismo oculocutáneo en Japón, de manera que es el tipo más frecuente de albinismo en personas japonesas con variantes identificables.[87] En un estudio de 22 personas de ascendencia italiana con variantes patogénicas de TYR, OCA2 o TYRP1 se identificaron 5 personas con variantes bialélicas de SLC45A2, 4 de ellos cumplieron con los criterios clínicos para un diagnóstico de albinismo oculocutáneo.[88] En conjunto se han identificado más de 600 variantes genéticas distintivas relacionadas con albinismo ocular.[89] Se piensa que el albinismo de tipo OCA4 es similar a otros tipos de albinismo, pero no se ha cuantificado el riesgo de CCE de piel en personas con estas variantes. En un estudio de 30 personas con OCA4 no se identificaron casos de cáncer de piel.[90] Cabe aclarar que en un metanálisis se observó que la variante SLC45A2 p.Phe374Leu fue protectora de melanoma, con una OR de 0,41 (IC 95 %, 0,33–0,50; P = 3,50 x 10-17).[91] Sin embargo, en este momento no se realizan pruebas de rutina en el ámbito clínico para estas variantes protectoras.
Se encontraron otros genes relacionados con albinismo oculocutáneo en un número pequeño de pacientes. El gen OCA5, ubicado en el cromosoma 4q24, se identificó en una familia de Pakistán, mientras que la causa de OCA6 son variantes patogénicas de SLC24A5 en el cromosoma 15q21.[92,93,94] Las variantes patogénicas de C10orf11 (LRMDA) causan OCA7, que se encontró en pacientes de las islas Faroe en Dinamarca.[95] Se han notificado pocos portadores de variantes patogénicas hasta el momento. Una mujer con OCA6 presentó queratosis actínica, pero se desconoce la incidencia del cáncer de piel en estas poblaciones.
Si bien la mayoría de los casos de albinismo oculocutáneo se heredan como un trastorno autosómico recesivo, en un estudio se encontró que hay sobrerrepresentación de variantes heterocigotas de genes como TYR, OCA2, TYRP1 y SLC45A2 en familias con múltiples casos de melanoma. Se requiere más investigación para determinar si estos genes son genes de susceptibilidad al melanoma de penetrancia moderada en heterocigotos.[96]
| Tipo | Subtipo | Gen | Población de notificación | Disponibilidad de la prueba clínica |
|---|---|---|---|---|
| Albinismo oculocutáneo de tipo 1 | 1A | TYR | Japonesa,[97]china,[98]blanca[99,100,101,102,103] | Sí |
| 1B | TYR | |||
| Albinismo oculocutáneo de tipo 2 | OCA2(genP) | Africana,[104,83,105]afroamericana,[106]indígena de los Estados Unidos[107] | Sí | |
| Albinismo oculocutáneo de tipo 3 | TYRP1 | Africana,[108] | Sí | |
| Albinismo oculocutáneo de tipo 4 | SLC45A2 (MATP) | Japonesa,[87]Italiana,[88]alemana[109] | Sí | |
| Albinismo oculocutáneo de tipo 5 | OCA5 | Pakistaní[92] | No en los Estados Unidos | |
| Albinismo oculocutáneo de tipo 6 | SLC24A5 | china,[93]Africana,[110]europea,[94]india[111] | Sí | |
| Albinismo oculocutáneo de tipo 7 | C10orf11(LRMDA) | de las islas Faroe,[95]danesa[95] | Sí |
Otros síndromes de albinismo
Un subgrupo de albinismo incluye a las personas que exhiben una tríada de albinismo, tiempo de sangrado prolongado y depósito de una sustancia ceroide en órganos como los pulmones y el aparato digestivo. Este síndrome, conocido como síndrome de Hermansky-Pudlak, se hereda de manera autosómica recesiva, pero es posible que tenga una herencia pseudodominante en familias puertorriqueñas, debido a la prevalencia elevada en esta población.[112] Se cree que la causa subyacente es un defecto en el transporte de melanosomas y lisosomas. Diversas variantes patogénicas ende locus diferentes se han vinculado con este síndrome, como HPS1, HPS3, HPS4, HPS5, HPS6, HPS7 (DTNBP1), HPS8 (BLOC1S3), y HPS9 (PLDN).[113,114,115,116,117,118,119,120] Las características pigmentarias varían bastante en este trastorno, en especial entre aquellos con variantes patogénicas de HPS1; algunos pacientes notifican aumento de la pigmentación de la piel y el cabello a medida que pasan los años. En una cohorte pequeña de pacientes con variantes del gen HPS1, 3 de 40 presentaron CCE y otros 3 presentaron CCB.[121] El síndrome de Hermansky-Pudlak de tipo 2, que incluye aumento de la susceptibilidad a infecciones debido a neutropenia congénita, se atribuye a defectos en AP3B1.[122]
Otros 2 síndromes se asocian con disminución de la pigmentación de la piel y los ojos. El síndrome de Chediak-Higashi es un trastorno autosómico recesivo que se caracteriza por cuerpos de inclusión eosinofílicos positivos para peroxidasa en precursores leucocitarios tempranos, hemofagocitosis, aumento de la susceptibilidad a infecciones y aumento de la incidencia de una linfohistiocitosis de fase acelerada. Las variantes patogénicas del gen LYST son la base de este síndrome, que suele producir la muerte en la primera década de vida.[123,124,125]
El síndrome de Griscelli, que también se hereda de manera autosómica recesiva, se describió originalmente como disminución de la pigmentación cutánea con hipomelanosis y deterioro neurológico, pero su presentación clínica es bastante variable. Esta combinación de síntomas ahora se denomina síndrome de Griscelli de tipo 1 o enfermedad de Elejalde. Se atribuye a las variantes patogénicas del gen MYO5A, que afecta el transporte de melanosomas.[126] Las personas con síndrome de Griscelli de tipo 2 exhiben disminución de la pigmentación cutánea e inmunodeficiencia pero no deterioro neurológico. También es posible que tengan hemofagocitosis o linfohistiocitosis que a menudo es mortal, como la que se observa en el síndrome de Chediak-Higashi. La causa del síndrome de Griscelli de tipo 2 son variantes patogénicas de RAB27A, gen que participa en la misma vía de transporte de melanosomas que MYO5A.[127] El síndrome de Griscelli de tipo 3 se manifiesta con hipomelanosis y no incluye trastornos neurológicos o inmunológicos. Las variantes patogénicas del gen de la melanofilina (MLPH) y MYO5A se relacionan con esta variante del síndrome de Griscelli.[128]
Epidermólisis ampollosa
Hay numerosas formas de epidermólisis ampollosa (EB), que se caracteriza por fisuras y ampollas en la piel. En un estudio del registro holandés de EB se encontró una incidencia general de EB de 41,3 por millón de personas nacidas vivas y de 22,4 por millón de personas de la población general.[129] De manera semejante, en un estudio alemán se encontró una incidencia de EB de 45,1 casos por millón de nacidos vivos.[130] No obstante, en un estudio de Inglaterra y Gales se encontró una incidencia de EB mucho más alta, de 67,8 casos por millón de habitantes.[131] En los casos más extremos, la EB produce la ausencia congénita de piel.[132] La EB distrófica y la EB de unión se relacionan con aumento del riesgo de cáncer de piel, en especial CCE.[133] Si bien este tipo de EB es difícil de identificar desde el punto de vista clínico, el uso de secuenciación de última generación permite detectar el subtipo de EB en hasta el 90 % de los casos.[134] El diagnóstico de EB se puede establecer mediante inmunofluorescencia o microscopía electrónica.[135] Los tipos de EB, las variantes patogénicas afectadas y las características fenotípicas se detallan en la siguiente revisión.[136] Para obtener más información, consultar la sección Epidermólisis ampollosa simple.
Epidermólisis ampollosa distrófica
Cerca del 95 % de las personas con el trastorno hereditario epidermólisis ampollosa distrófica (DEB) tienen una variante germinal patogénica detectable del gen COL7A1. Este gen (ubicado en 3p21.3), se expresa en los queratinocitos basales de la epidermis y codifica el colágeno de tipo VII. Este colágeno compone las fibrillas que unen la membrana basal con la dermis, y proporcionan estabilidad estructural y resistencia de la piel a traumatismos leves.[137] La falta de colágeno de tipo VII suele producir ampollas generalizadas en la piel (desde el nacimiento), atrofia y cicatrización cutánea.[137] Se puede consultar en inglés el registro internacional de variantes patogénicas de DEB en el siguiente enlace de Internet DEB Register.[138] En un estudio observacional se analizaron personas del registro holandés de EB por un periodo de 30 años. En los resultados se encontró que la DEB representa el 34,7 % de todos los diagnósticos de EB y que tiene una prevalencia puntual de 8,3 por millón de personas de la población general.[129] En estudios de los Países Bajos, Alemania, Inglaterra y Gales se observó que la incidencia de DEB osciló de 14,1 a 26,1 casos por millón de nacidos vivos.[129,130,131]
Hay dos subtipos de DEB de herencia recesiva: generalizada grave (RDEB-sev gen; antes conocida como de tipo Hallopeau-Siemens) y generalizada de otro tipo o generalizada intermedia (RDEB-O; antes conocida como de tipo no–Hallopeau-Siemens); y una forma de herencia dominante, epidermólisis ampollosa distrófica dominante (DDEB).[139] Estos síndromes son infrecuentes. La prevalencia por millón de personas en los Estados Unidos y la incidencia por millón de nacidos vivos son de 0,36 y 0,57 para la RDEB-sev gen, de 0,14 y 0,30 para la RDEB-O, y de 1,49 y 2,12 para la DDEB, respectivamente.[140] Las manifestaciones clínicas siguen una progresión de gravedad que complica el diagnóstico definitivo, en especial en niños muy pequeños. El subtipo generalizado grave, que se asocia con la formación de pseudosindactilia (malformación de manos en mitón por fusión membranosa interdigital) en la niñez temprana, acarrea un riesgo de CCE de hasta el 85 % a la edad de 45 años.[141,142] Estos cánceres aparecen en heridas que no cicatrizan y suelen metastatizar hasta causar la muerte en los 5 años posteriores al diagnóstico de CCE.[143] En una serie de casos, el CCE fue la principal causa de muerte en 15 pacientes con el subtipo generalizado grave.[144] La incidencia de CCE es más alta en el subtipo RDEB. En una revisión de 69 artículos en los que se incluyeron todos los tipos de EB, se identificaron 117 personas con CCE; 81 de estos casos (69,2 %) en personas con RDEB.[133] En este grupo, la mediana de edad en el momento del diagnóstico fue de 36 años (intervalo, 6–71 años). En un estudio se encontró que 18 de 283 (6,4 %) personas con RDEB presentaron un diagnóstico de CCE con una mediana de edad del diagnóstico de 22,6 años (intervalo intercuartílico, 20–27,7 años).[145] También se ha observado mortalidad temprana en este trastorno, con una tasa de mortalidad de hasta el 40 % a la edad de 30 años.[146] En un estudio epidemiológico de Alemania se encontró que la media de edad de muerte fue de 28,03 años en los pacientes con DEB.[130] Las manifestaciones extracutáneas de RDEB-sev gen incluyen estatura baja, anemia, estenosis del tubo digestivo y las vías urinarias, y cicatrización corneal que puede producir ceguera.
La tasa de variantes patogénicas de novo para el DDEB es de alrededor del 30 %; también se ha notificado mosaicismo germinal materno.[147,148] Las substituciones de glicina en los exones 73 a 75 son las variantes patogénicas de DDEB más comunes. G2034R y G2043R representan la mitad de estas variantes. Con menos frecuencia, las variantes patogénicas del sitio de empalme y las sustituciones de glicina y otros aminoácidos pueden originar una forma dominante de DEB. Por el contrario, se han descrito más de 400 variantes patogénicas para los dos tipos recesivos de EB. La forma recesiva de la enfermedad se debe principalmente a variantes nulas, aunque también se han notificado sustituciones de aminoácidos, variantes del sitio de corte y empalme y variantes de cambio de sentido. La omisión de un exón en el marco de lectura puede generar un proteína parcialmente funcional en la enfermedad recesiva. Una variante patogénica con efecto fundador, c.6527insC (p.R525X), se observó en 27 de 49 personas españolas con DEB recesiva.[149] Una variante patogénica con efecto fundador de COL7A1, pVal769LeuFsXI, se identificó en 11 de 15 familias en Sfax (sur de Túnez).[150] En 3 de 12 personas portadoras de por lo menos una copia de esta variante, se identificaron casos de CCE, entre ellos, 2 casos de inicio en la juventud a los 16 y 29 años. Las correlaciones entre genotipo y fenotipo indican una correlación inversa entre la cantidad de proteína funcional y la gravedad de la enfermedad.
Las variantes patogénicas de COL7A1 producen un enrollamiento helicoidal triple anormal con funcionamiento reducido, lo que aumenta la fragilidad cutánea y el ampollamiento. En los estudios de la carcinogénesis dirigida por RAS en queratinocitos de la RDEB generalizada grave, la retención del NC1 aminoterminal, el primer fragmento no colagenoso del colágeno de tipo VII, es oncogénico en ratones.[151] Es posible que la retención de esta secuencia medie interacciones entre el tumor y el estroma que promueven la carcinogénesis.
Epidermólisis ampollosa de la unión
La epidermólisis ampollosa de la unión (JEB) es un tipo de EB autosómica recesiva con una incidencia estimada de 2,68 por millón de personas nacidas vivas y una prevalencia estimada de 0,49 por millón de personas en los Estados Unidos.[140] En un estudio del registro holandés de EB se notificó una prevalencia estimada más alta con una tasa de 9,3 por millón de personas nacidas vivas y 2,1 por millón de personas en los Países Bajos.[129] En este estudio, alrededor del 19 % de todos los diagnósticos de EB correspondieron a JEB. La incidencia de JEB en Inglaterra y Gales (8,9 casos por millón nacidos vivos) fue similar a la de los Países Bajos y los Estados Unidos, pero en un estudio de Alemania se notificó una incidencia más alta de JEB (14,23 casos por millón nacidos vivos).[130,131] La JEB produce una tasa alta de mortalidad, alrededor del 50 % de los casos fallecen durante el primer año de vida.[152] Las variantes patogénicas de COL17A1 o en cualquiera de los genes codificadores de la laminina 332 y cualquiera de sus 3 subunidades, antes conocidas como laminina 5 (LAMA3, LAMB3, LAMC2), producen JEB.[153,154,155] Las personas con una forma grave de JEB, llamada de tipo Herlitz tienen un 18 % de riesgo acumulado de presentar CCE a los 25 años de edad.[156] En un estudio de COL17A1 en personas con un subtipo de JEB más leve, llamado JEB-otro tipo, se identificaron variantes patogénicas en 85 de 86 alelos de 43 participantes.[157] En este estudio, la ausencia absoluta de tinción para la proteína COL17A1 se asoció con un fenotipo de JEB grave.
Epidermodisplasia verruciforme
Las variantes patogénicas en cualquiera de dos genes adyacentes en el cromosoma 17q25 pueden causar epidermodisplasia verruciforme, un trastorno hereditario raro asociado con una mayor susceptibilidad a la infección por virus del papiloma humano (VPH). La infección con ciertos subtipos de VPH produce lesiones verrugosas generalizadas persistentes, que se convierten en CCE in situ e invasores en un 30 % a un 60 % de los pacientes.[158] Se piensa que la transformación maligna ocurre en cerca de la mitad de estas lesiones. Alrededor del 90 % de las lesiones se atribuyen a los tipos 5 y 8 de VPH,[159] aunque los tipos 14, 17, 20 y 47 a veces participan en estos casos. La relación entre la infección por el VPH y el aumento del riesgo de CCE también se demostró en personas sin epidermodisplasia verruciforme; en un estudio de casos y controles se observó que los anticuerpos contra VPH se encontraron con más frecuencia en el plasma de personas con CCE (OR, 1,6; IC 95 %, 1,2–2,3) que en el plasma de personas sin cáncer.[160]
Los genes asociados con este trastorno, EVER1 y EVER2, se identificaron en el 2002.[161] El patrón de herencia de estos genes es autosómico recesivo; sin embargo también se han notificado casos de herencia autosómica dominante.[162,163,164] Ambos productos de estos genes son proteínas transmembranas ubicadas en el retículo endoplasmático, y su función probable es la transducción de señales. Es probable que este efecto se produzca por regulación del equilibrio del cinc; se ha demostrado que estas proteínas forman un complejo con el transportador de cinc 1 (ZnT-1), que a su vez está bloqueado por ciertas proteínas de los VPH.[165]
En un estudio de casos y controles reciente se examinó el efecto de un polimorfismo específico en el gen EVER2 (rs7208422) sobre el riesgo de CCE cutáneo en 239 personas con antecedentes de CCE y 432 controles. Este polimorfismo es una variante de un solo nucleótido con codificación (A > T) en el exón 8, codón 306 del gen EVER2. La frecuencia del alelo T entre los controles fue de 0,45. La homocigosis para el polimorfismo causó un aumento modesto en el riesgo de CCE, con una OR ajustada de 1,7 (IC 95 %, 1,1–2,7) en relación con los homocigotos para el tipo normal del gen. En este estudio, también se encontró que aquellos con uno o más de los alelos T tienen mayor seropositividad para cualquier tipo de VPH y para los tipos de VPH 5 y 8, en comparación con el tipo normal.[166]
Hay evidencia que indica heterogeneidad no alélica en la epidermodisplasia verruciforme. En una persona nacida de progenitores emparentados con epidermodisplasia verruciforme, que además tenía infecciones bacterianas y fúngicas, se encontraron variantes patogénicas homocigotas R115X en el gen MST1.[167] Con análisis de ligamiento se identificó otro locus de susceptibilidad asociado con este trastorno en las regiones cromosómicas 2p21-p24 en una familia con vínculos de consanguinidad. A diferencia de las personas afectadas por variantes patogénicas de los genes EVER1 y EVER2, las personas afectadas por alteraciones en esta región genómica presentaban infecciones por el tipo 20 de VPH en lugar de los subtipos de VPH que suelen asociarse con este trastorno; esta familia no tenía antecedentes de CCE cutáneo.[168]
Anemia de Fanconi
La anemia de Fanconi es un trastorno complejo que se caracteriza por un aumento de la incidencia de tumores hematológicos y sólidos, como el CCE de piel. La anemia de Fanconi es una enfermedad de herencia autosómica recesiva. Se trata de un síndrome relativamente raro con una frecuencia estimada de portadores de 1 de cada 181 personas en los Estados Unidos (intervalo: 1 en 156 hasta 1 en 209) y una frecuencia de portadores de hasta 1 en 100 personas con ascendencia judía askenazí.[169] La leucemia es el tipo de cáncer más frecuente en esta población, pero también se han observado tasas elevadas de neoplasias gastrointestinales, de cabeza y cuello, y ginecológicas.[170] A la edad de 40 años, la anemia de Fanconi acarrea un riesgo del 8 % por año de presentar un tumor sólido;[170] la mediana de edad de diagnóstico de un tumor sólido es de 26 años.[171] Se han notificado múltiples casos de cáncer de encéfalo, mama, pulmón y riñón (tumor de Wilms) en esta población.[171] Hay pocos datos sobre la incidencia de NMSC en esta población; sin embargo en una revisión de la literatura se encontró que la edad de diagnóstico oscila entre los 25 y los 35 años, y que las mujeres se ven más afectadas que los hombres.[171,172,173,174,175]
Las personas con esta enfermedad tienen un aumento de la susceptibilidad a los agentes entrecruzadores del DNA (por ejemplo, mitomicina-C o diepoxibutano) y a la radiación ionizante y UV. El diagnóstico de esta enfermedad se determina al observar un aumento de la rotura cromosómica, reordenamientos o intercambios en las células después de la exposición a carcinógenos como el diepoxibutano.
Se han identificado 17 grupos de complementación en la anemia de Fanconi; la información detallada de los genes asociados a estos grupos se registra en el Cuadro 7.[176] Mediante secuenciación del exoma se identificó un subgrupo de personas que portan múltiples variantes patogénicas heterocigotas de los genes de la anemia de Fanconi,[177] lo que puede repercutir en la presentación fenotípica.
| Gen | Locus | Incidencia aproximada en pacientes con anemia de Fanconi (%) | Patrón de transmisión de la enfermedad |
|---|---|---|---|
| AR = autosómico recesivo; XLR =recesivo ligada al cromosoma X. | |||
| FANCA | 16q24.3 | 70 | AR |
| FANCB | Xp22.31 | Raro | XLR |
| FANCC | 9q22.3 | 10 | AR |
| FANCD1(BRCA2) | 13q12.3 | Raro | AR |
| FANCD2 | 3p25.3 | Raro | AR |
| FANCE | 6p21.3 | 10 | AR |
| FANCF | 11p15 | Raro | AR |
| FANCG(XRCC9) | 9p13 | 10 | AR |
| FANCI(KIAA1794) | 15q25-26 | Raro | AR |
| FANCJ(BACH1/BRIP1) | 17q22.3 | Raro | AR |
| FANCL(PHF9/POG) | 2p16.1 | Raro | AR |
| FANCM(Hef) | 14q21.3 | Raro | AR |
| FANCN(PALB2) | 16p12.1 | Raro | AR |
| FANCO(RAD51C) | 17q22 | Raro | AR |
| FANCP(SLX4/BTBD12) | 16p13.3 | Raro | AR |
| FANCQ(ERCC4/XPF) | 16p13.12 | Raro | AR |
| FANCS(BRCA1) | 17q21.31 | Raro | AR |
Las proteínas que participan en la reparación del entrecruzamiento del DNA se conocen como vía FANC por su participación en la anemia de Fanconi.[178] Estas proteínas interactúan con muchas otras proteínas vinculadas con riesgo de cáncer hereditario, entre ellas las del síndrome de Bloom y de la ataxia-telangiectasia. En investigaciones adicionales se determinó que FANCD1 es el mismo gen que BRCA2, un gen que causa predisposición al cáncer de mama y de ovario.[179] Se ha identificado que otros genes de la anemia de Fanconi, FANCJ (BRIP1) y FANCN (PALB2), también se comportan como genes de susceptibilidad, raros, al cáncer de mama.[180] Para obtener más información en inglés, consultar la sección Cancer Risks, Spectrum, and Characteristics en BRCA1 and BRCA2: Cancer Risks and Management, además de las secciones BRIP1, PALB2 y RAD51 en Genetics of Breast and Gynecologic Cancers. Las personas que son portadoras heterocigotas de otras variantes asociadas con la anemia de Fanconi no parecen tener aumento del riesgo de cáncer, excepto por un aumento del doble en la incidencia de cáncer de mama que se observa en portadoras de variantes patogénicas de FANCC.[181]
En 2018, un grupo reportó un aumento significativo en los casos de CCE (OR, 1,69; IC 95 %, 1,26–2,26) asociados con un alelo BRCA2 específico, que es relativamente prevalente en población islandesa (K3326X; frecuencia alélica, 1,1 %).[182] Este alelo conlleva la producción normal de una proteína alterada y los autores hipotetizaron que los portadores tienen aumento de la sensibilidad a factores ambientales, que exigen la reparación del DNA. Esta variante también se relacionó con un aumento en el riesgo de cáncer de pulmón de células pequeñas, cáncer de mama y cáncer de ovario (pero menor que el riesgo asociado con las variantes patogénicas de BRCA que disminuyen los niveles de proteína).
Disqueratosis congénita (síndrome de Zinsser-Cole-Engman)
La disqueratosis congénita, al igual que el síndrome de Werner, produce un envejecimiento prematuro y, por lo tanto, se considera una enfermedad progeroide. La tríada clínica clásica para el diagnóstico incluye distrofia ungueal, pigmentación reticular del tórax y el cuello y leucoplasia oral. Además, las personas con este trastorno tienen un riesgo mucho más alto de síndrome mielodisplásico, leucemia aguda e insuficiencia de la médula ósea. También se han descrito alteraciones oculares, dentales, neurológicas, gastrointestinales, pulmonares y esqueléticas; pero su expresión clínica es muy variable en esta enfermedad.[183] A veces también se presenta retraso del desarrollo en variantes de disqueratosis congénita, como el síndrome de Hoyeraal-Hreidarsson (HHS) y el síndrome de Revesz.
Cerca del 10 % de las personas con disqueratosis congénita presentaran tumores no hematológicos, a menudo antes de la tercera década de vida.[184,185] Los tumores sólidos quizás sean la primera manifestación de este trastorno. Los cánceres de cabeza y cuello fueron los que se notificaron con más frecuencia, lo que representó casi la mitad de los cánceres observados. El CCE cutáneo se presentó en cerca del 1,5 % de las personas, y la mediana de edad de diagnóstico fue de 21 años. Por lo general, estos cánceres se tratan como cualquier otro tipo de CCE de la piel.
Múltiples genes asociados con la función de los telómeros (DKC1, TERC, TINF2, NHP2, NOP10, RTEL1 y TERT) están implicados en la disqueratosis congénita; aproximadamente la mitad de los pacientes con diagnóstico clínico de esta enfermedad tienen variantes patogénicas identificadas en 1 de estos 7 genes.[186,187,188,189,190,191,192,193] Los genes TERC y TINF2 se heredan de manera autosómica dominante, mientras que NHP2 (NOLA2) y NOP10 (NOLA3) exhiben una herencia autosómica recesiva; RTEL1 y TERT se heredan de manera autosómica dominante o recesiva. Las variantes patogénicas de RTEL1 de herencia autosómica recesiva también se asocian con HHS.[194] En un estudio de más de 1000 personas de ascendencia judía asquenazí, se identificó una variante patogénica del sitio de empalme de RTEL1, c.3791G>A (p.R1264H), que tuvo una frecuencia de portadores del 1 % en población de ascendencia judía asquenazí ortodoxa y del 0,45 % en la población general de ascendencia judía asquenazí.[195]DKC1 exhibe un patrón de herencia recesiva ligada a X. Las alteraciones en estos genes producen acortamiento de los telómeros, que conlleva defectos en la proliferación y reordenamientos cromosómicos espontáneos.[196] Las concentraciones de TERC, el componente RNA del complejo de la telomerasa, son bajas en todos los pacientes con disqueratosis congénita.[197] Las variantes patogénicas de cambio de sentido del gen WRAP53, un gen con un producto proteínico que facilita el tráfico de telomerasas, también se han asociado con una forma autosómica recesiva de disqueratosis congénita.[198] Se identificaron variantes patogénicas de C16orf57 en 6 de 132 familias en quienes no se encontraron variantes de otros genes conocidos.[199] Las variantes patogénicas de C16orf57 también se asocian con poiquilodermia y neutropenia.[200] Para obtener más información sobre la poiquilodermia congénita, consultar la sección Síndrome de Rothmund-Thomson.
El abordaje recomendado para el diagnóstico comienza con un ensayo de panel de 6 células para analizar la longitud telomérica de los leucocitos. Si la longitud de los telómeros está en el 1 % más bajo en 3 o más tipos celulares, se indica el uso de pruebas genéticas moleculares.[201] Las pruebas de DKC1 a veces se realizan primero en probandos varones, porque las variantes patogénicas de este gen representan hasta el 36 % de las variantes que hasta el momento se han vinculado con disqueratosis congénita. Las variantes patogénicas de TINF2 y TERT son responsables del 11 % al 24 % y del 6 % al 10 % de los casos, respectivamente.[183,190,191,202,203]
Síndrome de Rothmund-Thomson
El síndrome de Rothmund-Thomson, también conocido como poiquilodermia congénita, es un trastorno hereditario caracterizado por inestabilidad cromosómica. La manifestación cutánea de esta afección es un exantema ampolloso eritematoso en la cara, los glúteos y las extremidades en la infancia temprana. Otras características de este síndrome son las telangiectasias, las anomalías esqueléticas, la estatura baja, las cataratas y el aumento del riesgo de osteosarcoma. Se desarrollan zonas de hiperpigmentación e hipopigmentación en la piel más adelante en la vida, y se puede presentar CCB o CCE a una edad temprana.[204] Se han notificado casos de múltiples CCE in situ que aparecieron en personas de solo 16 años.[205] No se ha caracterizado bien el aumento exacto del riesgo de cáncer de piel, pero la prevalencia puntual de los carcinomas de queratinocitos, incluyendo CCB y CCE, es del 2 % al 5 % en personas jóvenes afectadas por este síndrome.[206] Esta prevalencia claramente es más elevada que la del mismo grupo etario en la población general. Si bien se ha descrito un aumento en la sensibilidad a la radiación UV, los CCE también se encuentran en áreas de la piel que no están expuestas al sol.[207]
Una variante patogénica del gen RECQL4 se identifica en el 66 % de las personas afectadas con manifestaciones clínicas del síndrome de Rothmund-Thomson de tipo 2. Este gen se ubica en 8q24.3, y se cree que su herencia es autosómica recesiva. RECQL4 codifica la helicasa de DNA dependiente de ATP de tipo Q4, que facilita el desenrollamiento del DNA para permitir procesos celulares como la replicación, la transcripción y la reparación. También se ha indicado que esta proteína cumple una función en la reparación de rupturas de la cadena doble de DNA.[208] Otras variantes patogénicas de helicasas de DNA similares conllevan el desarrollo de trastornos hereditarios como el síndrome de Bloom y el síndrome de Werner.
Se han identificado por lo menos 19 variantes patogénicas truncadoras diferentes de este gen que son deletéreas.[209] Estas variantes patogénicas causan una disminución importante en los transcritos de RECQL4 en este subgrupo de personas con síndrome de Rothmund-Thomson.[210] Se ha encontrado que las células con deficiencia de RECQL4 son hipersensibles al estrés oxidativo, lo que produce disminución en la síntesis de DNA.[211] Las deficiencias en las helicasas de RecQ posibilitan la hiperrecombinación, lo que conlleva la pérdida de heterocigosidad. La pérdida de heterocigosidad relacionada con deficiencias de esta proteína indica que las helicasas son proteínas supresoras de tumores de tipo cuidadoras (caretaker).[212]
Se encontró que 3 de 6 familias con síndrome de Rothmund-Thomson de tipo 2 exhibían variantes patogénicas homocigotas del gen C16orf57. También se identificaron variantes patogénicas de este gen en personas con disqueratosis congénita y poiquilodermia con neutropenia, lo que indica un vínculo entre estos síndromes;[199,200] sin embargo, no se ha descrito bien el riesgo de cáncer de piel en estas afecciones. Para obtener más información, consultar la sección Disqueratosis congénita (síndrome de Zinsser-Engman-Cole).
El cuadro clínico inicial del síndrome de Rothmund-Thomson de tipo 1 es similar al tipo 2, excepto que las personas tienen cataratas juveniles bilaterales y no presentan osteosarcoma. En un estudio de 7 familias con 10 niños con características de síndrome de Rothmund-Thomson de tipo 1 se identificó una variante de empalme intrónica profunda del gen ANAPC1.[213] De las personas analizadas, 5 eran homocigotas para esta variante, y 3 personas presentaron una segunda variante patogénica predicha de ANAPC1 en trans. La variante de empalme llevó a la introducción de un nuevo exón, que causó un codón de terminación prematuro y una descomposición subsiguiente de los transcritos de ANAPC1 mediada por el cambio en la secuencia (sin sentido) que conllevó a reducción de las concentraciones de la proteína. ANAPC1 es un miembro del complejo promotor de la anafase o ciclosoma.
Síndrome de Bloom
La pérdida de estabilidad genómica es una causa importante del síndrome de Bloom. En este trastorno se produce aumento de la ruptura cromosómica y se diagnostica mediante un aumento de los intercambios de cromátidas hermanas en el análisis cromosómico. Las manifestaciones clínicas del síndrome de Bloom incluyen retraso grave del crecimiento, infecciones recurrentes, diabetes, enfermedad pulmonar crónica y aumento de la susceptibilidad a muchos tipos de cáncer. En un estudio de 290 participantes del Bloom Syndrome Registry (inscritos desde 1960 hasta 2021), se encontró que 155 participantes (53 %) presentaron una o más neoplasias malignas; la incidencia cumulada de cualquier tipo de cáncer a los 40 años fue del 83 %.[214] La mediana de supervivencia fue de 36,2 años. La mediana de supervivencia varió para aquellos con diagnóstico de una neoplasia maligna (25,4 años) y aquellos con diagnóstico de un tumor sólido (37,7 años). La mediana de supervivencia no se ajustó por edad en el momento del diagnóstico de cáncer.[214]
El cáncer de piel explica alrededor del 9 % de los tumores en el Bloom Syndrome Registry.[215] Los cánceres de piel se presentan a una edad temprana en esta población, la media de edad en el momento del diagnóstico es de 31 a 33 años.[214,215] Casi el 12 % de los participantes del registro presentan cáncer de piel, el más común es CCB.[214] La media de edad en el momento del diagnóstico de cáncer de piel fue de 33 años. La lesión cutánea típica que se observa en este trastorno es una erupción telangiectásica eritematosa fotosensible que se presenta en el primer o segundo año de vida.[215] Aunque esta erupción se encuentra con mayor frecuencia en la cara, también se observa en el dorso de las manos o los antebrazos. El CCE cutáneo es la tercera neoplasia maligna más común relacionada con este trastorno.
El gen BLM, ubicado en el brazo corto del cromosoma 15, es el único gen que se conoce está relacionado con el síndrome de Bloom. Este gen codifica una proteína de 1417 aminoácidos regulada por el ciclo celular que actúa como ATPasa dependiente de DNA y también desenrolla la doble hélice de DNA. El dominio de helicasa exhibe semejanza considerable con la subfamilia RecQ de las helicasas de DNA. Se piensa que la ausencia de este producto de gen desestabiliza otras enzimas que participan en la replicación y reparación del DNA.[216,217]
Este raro síndrome hereditario de ruptura cromosómica se hereda de manera autosómica recesiva y se caracteriza por pérdida de la estabilidad genómica. Se han descrito 64 variantes patogénicas del gen BLM que abarcan inserciones y deleciones de nucleótidos (41 %), variantes sin sentido (30 %), variantes que producen errores de empalme (14 %) y variantes de cambio de sentido (16 %).[218,219] Una variante patogénica específica identificada en la población judía asquenazí es una deleción de 6-bp/inserción de 7-bp en el nucleótido 2281, que se designa como BLMASH.[220] Muchas de estas variantes producen el truncamiento del extremo C de la proteína, que impide la ubicación normal de esta proteína en el núcleo. La ausencia de la proteína BLM funcional ocasiona aumento de las tasas de variantes patogénicas y recombinación. Esta hipermutabilidad somática conduce a un aumento del riesgo de cáncer a una edad temprana en prácticamente todos los órganos, incluso la piel.
Se encontró que las células de personas con síndrome de Bloom tienen respuestas anormales a la radiación UV. La acumulación nuclear normal de TP53 después de la radiación UV estaba ausente en 2 de 11 cultivos celulares primarios de personas con síndrome de Bloom; en contraste, las respuestas en cultivos de personas que tienen XP y ataxia-telangiectasia eran normales.[221] También se encontró que el producto del gen BLM formó complejos con las proteínas de Fanconi, por lo que se planteó la posibilidad de conexiones entre las vías de BLM y de la anemia de Fanconi en la estabilidad del DNA.[222]
Síndrome de Werner
Al igual que el síndrome de Bloom, el síndrome de Werner se caracteriza por un inestabilidad cromosómica espontánea, lo que aumenta la susceptibilidad al cáncer y el envejecimiento prematuro. Los criterios diagnósticos, a menudo en el entorno de consanguinidad, incluyen cataratas, estatura corta, encanecimiento prematuro o adelgazamiento del cabello, y una prueba positiva de ácido hialurónico en orina de 24 horas. Las manifestaciones cutáneas cardinales de este trastorno son cambios cutáneos esclerodermatosos, ulceraciones, atrofia y cambios en la pigmentación. Las personas con este síndrome tienen una esperanza de vida promedio inferior a 50 años.[223] Los cánceres en este síndrome son de inicio temprano y se presentan hasta en el 43 % de estos pacientes.[224] Se ha descrito una variedad de tumores asociados con este trastorno en la población japonesa, entre ellos, aumento en la incidencia de sarcoma, cáncer de tiroides y cáncer de piel.[225] Alrededor del 20 % de los cánceres que se notifican en este síndrome son cutáneos, el melanoma y el CCE de la piel explican el 14 % y el 5 % de los casos, respectivamente.[226] En un estudio de 189 personas con síndrome de Werner, el riesgo estimado de melanoma estaba elevado hasta 53 veces en estas personas.[227] El CCE se diagnosticó con menos frecuencia. Hay predominio de melanomas lentiginosos acrales y a veces los CCE presentan un comportamiento más agresivo con metástasis a ganglios linfáticos y órganos internos.[225,228]
Se han identificado variantes patogénicas del gen WRN en el cromosoma 8p12-p11.2 en cerca del 90 % de las personas con este síndrome; no se sabe que otros genes estén relacionados con el síndrome de Werner.[224,229,230,231,232] Se cree que la herencia de este gen es autosómica recesiva. El producto del gen WRN es una proteína multifuncional que incluye una exonucleasa de DNA y una helicasa de DNA dependiente de ATP perteneciente a la subfamilia RecQ. Esta proteína quizás cumpla una función en procesos como la reparación del DNA, la recombinación, la replicación, la transcripción y diversas funciones de DNA.[233,234,235,236,237,238,239,240,241] La disfunción de los telómeros se ha asociado con envejecimiento prematuro y susceptibilidad al cáncer.[242] En otros síndromes de inestabilidad cromosómica, se encuentran alteradas otras helicasas con funciones similares, como BLM en el síndrome de Bloom y RecQL4 en el síndrome de Rothmund-Thomson.
Las variantes patogénicas descritas del gen WRN incluyen todos los tipos de variantes, como las de cambio de sentido, de sitio de empalme y reordenamientos grandes.[243] Se han encontrado unas pocas variantes patogénicas con efecto fundador que incluyen una variante del sitio de empalme (antes conocida como IVS 25-1G→C; que ahora se conoce como c.3139-1G>C) en más del 50 % de las personas japonesas afectadas.[244,245]
Las variantes patogénicas del gen WRN causan disminución en la concentración nuclear del producto génico. Las concentraciones intracelulares de mRNA (ARN mensajero) y de la proteína que se origina de la variante también se reducen de manera marcada en comparación con las de tipo normal. La semivida de los mRNA y de la proteína que se origina de la variante también es más corta que la de los mRNA y proteínas de tipo normal.[246,247]
MC1R
En un metanálisis se observó que a mayor número de variantes patogénicas de MC1R en el portador, mayor es el riesgo de presentar CCE y CCB. Las personas con 2 o más variantes patogénicas de MC1R presentaron una OR de 2,48 (IC 95 %, 1,96–3,15) para el CCB y una OR de 2,80 (IC 95 %, 1,71–4,57) para el CCE; este riesgo aumentó en personas de cabello rojo.[248] En un estudio de personas con diagnóstico de CCB antes de los 40 años también se encontró una asociación más fuerte entre el CCB y las variantes patogénicas de MC1R en personas con características fenotípicas que no suelen considerarse de riesgo alto. Para obtener más información, consultar la sección MC1R en la sección Melanoma.[249]
Intervenciones
Prevención y tratamiento de los cánceres de piel
En un ensayo clínico controlado con placebo y con enmascaramiento doble de fase III realizado en Australia, se evaluaron los efectos de la nicotinamida oral (vitamina B3) en 386 personas con antecedente de por lo menos 2 cánceres de piel no melanoma en los 5 años previos a la inscripción en el estudio.[250] La media de edad de los participantes del estudio fue de 66,4 años, y el 63 % eran hombres. Al cabo de 12 meses de tratamiento, aquellos que tomaban nicotinamida 500 mg 2 veces al día exhibieron una reducción del 30 % en la incidencia de CCB nuevos (IC 95 %, 0–51 %; P = 0,05). También se observó una reducción estadísticamente significativa en las queratosis actínicas, las lesiones cutáneas precursoras de los CCE. La tasa de nuevos cánceres de piel no melanoma fue un 23 % más baja en el grupo de nicotinamida (IC 95 %, 4–38 %; P = 0,02) que en el grupo de placebo. No se observaron diferencias clínicas significativas en los efectos adversos entre los dos grupos, y no hubo evidencia de beneficio después de la interrupción del tratamiento con nicotinamida. Cabe destacar que este estudio no se llevó a cabo en una población con una predisposición genética al CCB identificada, pero se llevó a cabo en una parte del mundo con incidencia alta de cáncer de piel causado por la luz solar.
Nivel de evidencia (nicotinamida): 1aii
Debido a que muchos de los síndromes descritos antes son raros, se han llevado a cabo pocos ensayos clínicos en estas poblaciones específicas. Sin embargo, se ha obtenido información valiosa a partir de la experiencia clínica relacionada con el riesgo del cáncer de piel y el tratamiento en la población con XP. La evasión estricta de la exposición al sol desde la infancia, el uso de ropa protectora y la vigilancia clínica de la piel son componentes esenciales del tratamiento de la XP. Se recomienda tomar fotografías de toda la piel, las conjuntivas y los párpados como ayuda durante el seguimiento clínico.[251] Aunque se han realizado pocos estudios sobre el tratamiento del CCE en la población con XP, en la mayoría de los casos el tratamiento es similar al recomendado para la población general. Las queratosis actínicas se tratan con terapias tópicas como fluorouracilo (5-FU), crioterapia con nitrógeno líquido o dermoabrasión, mientras que los cánceres cutáneos por lo general se tratan con métodos quirúrgicos.[252]
Nivel de evidencia: 5
La isotretinoína oral se ha utilizado como quimioprevención en pacientes con XP y los resultados son prometedores. En un pequeño estudio sobre el uso diario de isotretinoína (ácido retinoico 13-cis administrado en dosis de 2 mg/kg/día), se redujo la incidencia de NMSC en un 63 % en un número pequeño de personas con XP. Los efectos tóxicos del tratamiento abarcan síntomas mucocutáneos, alteraciones en las pruebas del funcionamiento hepático y las concentraciones de triglicéridos, además de síntomas osteomusculares como artralgias, calcificaciones de tendones y ligamentos, y osteoporosis.[253,254] La disminución de la dosis a 0,5 mg/kg/día redujo los efectos tóxicos y la frecuencia del cáncer de piel en 3 de 7 personas (43 %); el aumento de la dosis a 1 mg/kg/día produjo disminución de la frecuencia del cáncer de piel en 3 de las 4 personas que no respondieron a la dosis más baja.[255] La isotretinoína oral quizás sea útil como quimioprevención en otros síndromes de cáncer de piel hereditario, como el síndrome de nevo de células basales (SNCB), el síndrome de Rombo, la EB y la epidermodisplasia verruciforme.[256,257]
Nivel de evidencia (isotretinoína oral para la XP): 3aii
Nivel de evidencia (isotretinoína oral para el SNCB, síndrome de Rombo y epidermodisplasia verruciforme): 5
En un ensayo aleatorizado controlado con placebo en 30 pacientes con XP se investigó el efecto quimiopreventivo de la loción liposomal T4N5 para uso tópico que contiene la enzima bacteriana T4 endonucleasa V.[258] Aunque no se observó un efecto en la incidencia del CCE, se identificaron 17,7 menos queratosis actínicas por año en el grupo de tratamiento. Además, se observaron 1,6 menos casos de CCB al año en los pacientes que recibieron esta terapia. Ambos resultados fueron estadísticamente significativos. El riesgo de CCB se redujo en un 47 %, lo que se considera en el límite de significación estadística. No se notificaron efectos adversos farmacológicos significativos. Hasta la fecha, la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA) no ha autorizado el uso de este producto.
Nivel de evidencia: 1aii
Se ha investigado la terapia con 5-FU en pacientes con XP que presentan un CCE irresecable. En un estudio prospectivo se usaron varios métodos de tratamiento, como terapia tópica de las lesiones, infusión sistémica breve de ácido fólico e infusión sistémica continua de ácido fólico con cisplatino. El tratamiento tópico con 5-FU demostró un poco de eficacia, pero en algunos casos quedó tumor viable en la profundidad de la dermis. La quimioterapia sistémica produjo 1 respuesta completa y 3 respuestas parciales en 5 pacientes, lo que indica que es una opción de tratamiento para lesiones generalizadas.[259] En esta población se ha recomendado la reducción de la dosis de quimioterapéuticos sistémicos en un 30 % a un 50 % por la sensibilidad elevada de las células de XP.[260]
Nivel de evidencia: 3diii
En los pacientes con EB, el tratamiento preferido sigue siendo la escisión local amplia del CCE con márgenes de 2 cm. Según el tamaño del CCE, es posible que sea difícil evaluar a estos pacientes. La eliminación completa de los tumores del CCE a veces exige una cirugía de control continuo de márgenes (escisión de Mohs) o el uso de márgenes quirúrgicos amplios y es posible que hagan falta múltiples intervenciones quirúrgicas. En ocasiones se necesita reorganizar los tejidos o usar sustitutos dérmicos para lograr el cierre de la herida.[261] Por otra parte, es posible que se considere la amputación como una opción para reducir la recidiva de la enfermedad, aunque no está clara la repercusión en la supervivencia. No se ha determinado la función de la biopsia de ganglio linfático centinela en esta población.[257]
En las directrices vigentes se recomienda que las personas con EB que tienen CCE irresecables reciban radioterapia, pero es posible que se necesite administrar la dosis en fracciones más pequeñas para disminuir el riesgo de descamación cutánea. También es posible considerar el uso de antagonistas de receptor del factor de crecimiento epidérmico o inhibidores de tirosina–cinasas en personas con CCE avanzado.[257]
Nivel de evidencia: 5
No hay muchos datos sobre personas con trastornos genéticos distintos al XP, pero las medidas generales de protección solar siguen siendo importantes. La protección cuidadosa de la piel y los ojos es la base de la prevención en todos los pacientes con aumento de la susceptibilidad al cáncer de piel. Los puntos clave son evasión de la exposición al sol en horas pico, la ropa y los lentes de protección, además del uso atento de protector solar. También se ha recomendado que algunos grupos con síndromes de cáncer de piel hereditario, como la epidermodisplasia verruciforme, eviten la radioterapia.[158] Sin embargo, los pacientes con XP y cánceres de piel irresecables o cánceres internos, como astrocitoma de médula espinal o glioblastomas en el encéfalo, han reaccionado bien al tratamiento con dosis terapéuticas estándar de radioterapia.[53] Algunos expertos recomiendan una evaluación dermatológica cada 6 meses y una evaluación oftalmológica cada año en estas poblaciones de riesgo alto. En las directrices para el tratamiento de los pacientes con EB se recomiendan exploraciones cutáneas cada 3 a 6 meses desde los 10 años de edad en personas con el subtipo RDEB-sev gen de la enfermedad.[257] Para las personas con otros subtipos de EB, se recomiendan exploraciones cutáneas cada 6 a 12 meses desde los 20 años, cuando no se ha establecido un diagnóstico de CCE. En esta población también se recomienda un examen dental cada 6 meses.[257]
Nivel de evidencia: 5
Para las personas con DEB, el cuidado de heridas es fundamental. El uso de crema de sulfadiacina de plata, miel de grado médico y apósitos blandos de silicona puede ser útil en estos casos. Otro aspecto fundamental que se debe considerar para la cicatrización de las heridas en estos pacientes es conservar el estado nutricional, que se puede afectar debido a estenosis esofágicas, anemia por deficiencia de hierro, infecciones e inflamación. Es posible que se justifique la administración de multivitaminas, a menudo en dosis más altas que las recomendadas para la población general.[262]
Nivel de evidencia: 5
El mantenimiento de la integridad de la piel es un factor clave para el tratamiento de los pacientes con JEB. En un pequeño estudio de 5 lactantes con JEB generalizada grave con al menos una variante patogénica de LAMB3 que produce truncamiento prematuro, se evaluaron los efectos clínicos del tratamiento con gentamicina (7,5 mg/kg/día durante 3 semanas) en la fragilidad cutánea y otras características clínicas. El tratamiento no afectó la mortalidad general, pero hubo una disminución de la fragilidad cutánea en 4 de los 5 pacientes; una mejora de la percepción de calidad de vida según lo notificado por los cuidadores, incluso un aumento de la actividad física sin ampollas; y un aumento de la expresión de la laminina 332 en la piel.[263]
Nivel de evidencia: 3c
Se ha explorado el trasplante de médula ósea para pacientes con DEB; sin embargo, no hay evidencia de que esta intervención disminuya el cáncer de piel.[264] Se llevó a cabo un ensayo aleatorizado controlado con placebo y con enmascaramiento doble de infusión de células madre de médula ósea no hematopoyéticas con ciclosporina o sin esta en 14 pacientes con DEB recesiva. La justificación de este estudio fue que las células madre mesenquimatosas (CSM) tienen el potencial de diferenciarse en fibroblastos dérmicos, la célula que más expresa colágeno de tipo VII. Se asignó al azar a 7 pacientes para recibir CSM con 5 mg/kg/día de ciclosporina y otros 7 pacientes recibieron solo CSM. El número de nuevas ampollas y la tasa de curación de las ampollas cutáneas mejoró de manera significativa en ambos grupos (P = 0,003 para el número de nuevas ampollas en el grupo de terapia combinada y P = 0,004 en el grupo que recibió solo CSM; P < 0,001 para la tasa de curación de ampollas cutáneas en ambos grupos). Sin embargo, no se observó diferencia entre los grupos.[265]
Nivel de evidencia (CSM para la prevención de ampollas): 1b
Nivel de evidencia (CSM para el tratamiento de ampollas): 1
Tratamientos futuros para la epidermólisis ampollosa
Los equipos de investigadores están aprovechando los avances recientes en tecnologías para estudiar las estrategias nuevas de tratamiento de la EB dominante y recesiva.[266,267,268,269] La técnica del sistema compuesto de la endonucleasa 9 asociada a grupos de repeticiones palindrómicas cortas en intervalos regulares (CRISPR)-(Cas9) se usa para editar el DNA. Un grupo de investigación usó CRISPR/Cas9 para corregir una variante patogénica hereditaria de COL7A1 en queratinocitos aislados de un paciente con RDEB.[266] Los queratinocitos que contenían la versión correcta de COL7A1 se trasplantaron de manera exitosa a ratones y la tinción de los injertos de piel mostró una piel normal. En otro estudio se usó un método diferente, una infección por retrovirus, para introducir un COL7A1 normal en los queratinocitos de 4 pacientes con RDEB.[268] Los queratinocitos regularizados se ensamblaron a las láminas del injerto epidérmico y se trasplantaron a 6 áreas de heridas en cada uno de los 4 pacientes del estudio. Los injertos fueron bien tolerados y, en un estudio posterior, se encontró mayor capacidad de curación que la piel sin corregir. En un ensayo prospectivo pequeño de gentamicina intravenosa en pacientes pediátricos con EB de unión, se observó que mejoró la cicatrización de la herida y la calidad de vida después del tratamiento.[270] Todas estas terapias todavía se encuentran en etapas iniciales de investigación y no se han evaluado en ensayos clínicos controlados aleatorizados.
En un ensayo aleatorizado de 31 pacientes con DEB se comparó el uso tópico de beremagene geperpavec con un placebo. El beremagene geperpavec es una terapia génica tópica a base del virus del herpes simplex de tipo 1 (VSH-1) que transporta el gen COL7A1 a la piel y por lo tanto, restaura la producción de la proteína colágeno VII. Esta terapia mejoró la cicatrización de heridas y redujo la intensidad del dolor durante el cambio de vendaje.[271]
Nivel de evidencia: 1
Referencias:
- Rogers HW, Weinstock MA, Harris AR, et al.: Incidence estimate of nonmelanoma skin cancer in the United States, 2006. Arch Dermatol 146 (3): 283-7, 2010.
- American Cancer Society: Cancer Facts and Figures 2024. American Cancer Society, 2024. Available online. Last accessed June 21, 2024.
- Rogers HW, Weinstock MA, Feldman SR, et al.: Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol 151 (10): 1081-6, 2015.
- Flohil SC, van der Leest RJ, Arends LR, et al.: Risk of subsequent cutaneous malignancy in patients with prior keratinocyte carcinoma: a systematic review and meta-analysis. Eur J Cancer 49 (10): 2365-75, 2013.
- Armstrong BK, Kricker A: The epidemiology of UV induced skin cancer. J Photochem Photobiol B 63 (1-3): 8-18, 2001.
- Rosso S, Zanetti R, Martinez C, et al.: The multicentre south European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 73 (11): 1447-54, 1996.
- Gallagher RP, Hill GB, Bajdik CD, et al.: Sunlight exposure, pigmentation factors, and risk of nonmelanocytic skin cancer. II. Squamous cell carcinoma. Arch Dermatol 131 (2): 164-9, 1995.
- Lindelöf B, Sigurgeirsson B, Tegner E, et al.: PUVA and cancer risk: the Swedish follow-up study. Br J Dermatol 141 (1): 108-12, 1999.
- Lim JL, Stern RS: High levels of ultraviolet B exposure increase the risk of non-melanoma skin cancer in psoralen and ultraviolet A-treated patients. J Invest Dermatol 124 (3): 505-13, 2005.
- Karagas MR, Stannard VA, Mott LA, et al.: Use of tanning devices and risk of basal cell and squamous cell skin cancers. J Natl Cancer Inst 94 (3): 224-6, 2002.
- Guo X, Fujino Y, Ye X, et al.: Association between multi-level inorganic arsenic exposure from drinking water and skin lesions in China. Int J Environ Res Public Health 3 (3): 262-7, 2006.
- Chen Y, Hall M, Graziano JH, et al.: A prospective study of blood selenium levels and the risk of arsenic-related premalignant skin lesions. Cancer Epidemiol Biomarkers Prev 16 (2): 207-13, 2007.
- Karagas MR, Stukel TA, Morris JS, et al.: Skin cancer risk in relation to toenail arsenic concentrations in a US population-based case-control study. Am J Epidemiol 153 (6): 559-65, 2001.
- Schwartz RA: Arsenic and the skin. Int J Dermatol 36 (4): 241-50, 1997.
- Mehta S, Goyal U, Gupta LK, et al.: Chronic Arsenicosis: Cases from a Nonendemic Area of South Rajasthan. Indian J Dermatol 64 (2): 164, 2019 Mar-Apr.
- Das KK, Chakaraborty A, Rahman A, et al.: Incidences of malignancy in chronic burn scar ulcers: experience from Bangladesh. Burns 41 (6): 1315-21, 2015.
- Koh D, Wang H, Lee J, et al.: Basal cell carcinoma, squamous cell carcinoma and melanoma of the skin: analysis of the Singapore Cancer Registry data 1968-97. Br J Dermatol 148 (6): 1161-6, 2003.
- Asgari MM, Warton EM, Whittemore AS: Family history of skin cancer is associated with increased risk of cutaneous squamous cell carcinoma. Dermatol Surg 41 (4): 481-6, 2015.
- Halder RM, Bang KM: Skin cancer in blacks in the United States. Dermatol Clin 6 (3): 397-405, 1988.
- Asuquo ME, Ebughe G: Major dermatological malignancies encountered in the University of Calabar Teaching Hospital, Calabar, southern Nigeria. Int J Dermatol 51 (Suppl 1): 32-6, 36-40, 2012.
- English DR, Armstrong BK, Kricker A, et al.: Demographic characteristics, pigmentary and cutaneous risk factors for squamous cell carcinoma of the skin: a case-control study. Int J Cancer 76 (5): 628-34, 1998.
- Kricker A, Armstrong BK, English DR, et al.: Pigmentary and cutaneous risk factors for non-melanocytic skin cancer--a case-control study. Int J Cancer 48 (5): 650-62, 1991.
- Akgüner M, Barutçu A, Yilmaz M, et al.: Marjolin's ulcer and chronic burn scarring. J Wound Care 7 (3): 121-2, 1998.
- Friedman R, Hanson S, Goldberg LH: Squamous cell carcinoma arising in a Leishmania scar. Dermatol Surg 29 (11): 1148-9, 2003.
- Jensen P, Hansen S, Møller B, et al.: Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol 40 (2 Pt 1): 177-86, 1999.
- Hartevelt MM, Bavinck JN, Kootte AM, et al.: Incidence of skin cancer after renal transplantation in The Netherlands. Transplantation 49 (3): 506-9, 1990.
- Lindelöf B, Sigurgeirsson B, Gäbel H, et al.: Incidence of skin cancer in 5356 patients following organ transplantation. Br J Dermatol 143 (3): 513-9, 2000.
- Krynitz B, Edgren G, Lindelöf B, et al.: Risk of skin cancer and other malignancies in kidney, liver, heart and lung transplant recipients 1970 to 2008--a Swedish population-based study. Int J Cancer 132 (6): 1429-38, 2013.
- Glover MT, Niranjan N, Kwan JT, et al.: Non-melanoma skin cancer in renal transplant recipients: the extent of the problem and a strategy for management. Br J Plast Surg 47 (2): 86-9, 1994.
- Kaplan AL, Cook JL: Cutaneous squamous cell carcinoma in patients with chronic lymphocytic leukemia. Skinmed 4 (5): 300-4, 2005 Sep-Oct.
- Euvrard S, Kanitakis J, Decullier E, et al.: Subsequent skin cancers in kidney and heart transplant recipients after the first squamous cell carcinoma. Transplantation 81 (8): 1093-100, 2006.
- Herrero JI, España A, D'Avola D, et al.: Subsequent nonmelanoma skin cancer after liver transplantation. Transplant Proc 44 (6): 1568-70, 2012 Jul-Aug.
- Cantwell MM, Murray LJ, Catney D, et al.: Second primary cancers in patients with skin cancer: a population-based study in Northern Ireland. Br J Cancer 100 (1): 174-7, 2009.
- Epstein E: Value of follow-up after treatment of basal cell carcinoma. Arch Dermatol 108 (6): 798-800, 1973.
- Møller R, Nielsen A, Reymann F: Multiple basal cell carcinoma and internal malignant tumors. Arch Dermatol 111 (5): 584-5, 1975.
- Bergstresser PR, Halprin KM: Multiple sequential skin cancers. The risk of skin cancer in patients with previous skin cancer. Arch Dermatol 111 (8): 995-6, 1975.
- Robinson JK: Risk of developing another basal cell carcinoma. A 5-year prospective study. Cancer 60 (1): 118-20, 1987.
- Greenberg ER, Baron JA, Stukel TA, et al.: A clinical trial of beta carotene to prevent basal-cell and squamous-cell cancers of the skin. The Skin Cancer Prevention Study Group. N Engl J Med 323 (12): 789-95, 1990.
- Karagas MR, Stukel TA, Greenberg ER, et al.: Risk of subsequent basal cell carcinoma and squamous cell carcinoma of the skin among patients with prior skin cancer. Skin Cancer Prevention Study Group. JAMA 267 (24): 3305-10, 1992.
- Helgadottir H, Höiom V, Tuominen R, et al.: CDKN2a mutation-negative melanoma families have increased risk exclusively for skin cancers but not for other malignancies. Int J Cancer 137 (9): 2220-6, 2015.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Hussain SK, Sundquist J, Hemminki K: The effect of having an affected parent or sibling on invasive and in situ skin cancer risk in Sweden. J Invest Dermatol 129 (9): 2142-7, 2009.
- Hemminki K, Zhang H, Czene K: Familial invasive and in situ squamous cell carcinoma of the skin. Br J Cancer 88 (9): 1375-80, 2003.
- Lindström LS, Yip B, Lichtenstein P, et al.: Etiology of familial aggregation in melanoma and squamous cell carcinoma of the skin. Cancer Epidemiol Biomarkers Prev 16 (8): 1639-43, 2007.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Loh AYT, Špoljar S, Neo GYW, et al.: Huriez syndrome: Additional pathogenic variants supporting allelism to SMARCAD syndrome. Am J Med Genet A 188 (6): 1752-1760, 2022.
- DiGiovanna JJ, Kraemer KH: Shining a light on xeroderma pigmentosum. J Invest Dermatol 132 (3 Pt 2): 785-96, 2012.
- Kraemer KH, Lee MM, Andrews AD, et al.: The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 130 (8): 1018-21, 1994.
- Bradford PT, Goldstein AM, Tamura D, et al.: Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48 (3): 168-76, 2011.
- Moussaid L, Benchikhi H, Boukind EH, et al.: [Cutaneous tumors during xeroderma pigmentosum in Morocco: study of 120 patients] Ann Dermatol Venereol 131 (1 Pt 1): 29-33, 2004.
- Brooks BP, Thompson AH, Bishop RJ, et al.: Ocular manifestations of xeroderma pigmentosum: long-term follow-up highlights the role of DNA repair in protection from sun damage. Ophthalmology 120 (7): 1324-36, 2013.
- Merideth M, Tamura D, Angra D, et al.: Reproductive Health in Xeroderma Pigmentosum: Features of Premature Aging. Obstet Gynecol 134 (4): 814-819, 2019.
- DiGiovanna JJ, Patronas N, Katz D, et al.: Xeroderma pigmentosum: spinal cord astrocytoma with 9-year survival after radiation and isotretinoin therapy. J Cutan Med Surg 2 (3): 153-8, 1998.
- Nikolaev S, Yurchenko AA, Sarasin A: Increased risk of internal tumors in DNA repair-deficient xeroderma pigmentosum patients: analysis of four international cohorts. Orphanet J Rare Dis 17 (1): 104, 2022.
- Robbins JH: Xeroderma pigmentosum complementation group H is withdrawn and reassigned to group D. Hum Genet 88 (2): 242, 1991.
- Khan SG, Oh KS, Shahlavi T, et al.: Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 27 (1): 84-94, 2006.
- Hirai Y, Noda A, Kodama Y, et al.: Increased risk of skin cancer in Japanese heterozygotes of xeroderma pigmentosum group A. J Hum Genet 63 (11): 1181-1184, 2018.
- Messaoud O, Ben Rekaya M, Cherif W, et al.: Genetic homogeneity of mutational spectrum of group-A xeroderma pigmentosum in Tunisian patients. Int J Dermatol 49 (5): 544-8, 2010.
- Ben Rekaya M, Messaoud O, Talmoudi F, et al.: High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet 54 (7): 426-9, 2009.
- Cartault F, Nava C, Malbrunot AC, et al.: A new XPC gene splicing mutation has lead to the highest worldwide prevalence of xeroderma pigmentosum in black Mahori patients. DNA Repair (Amst) 10 (6): 577-85, 2011.
- Doubaj Y, Laarabi FZ, Elalaoui SC, et al.: Carrier frequency of the recurrent mutation c.1643_1644delTG in the XPC gene and birth prevalence of the xeroderma pigmentosum in Morocco. J Dermatol 39 (4): 382-4, 2012.
- Hirai Y, Kodama Y, Moriwaki S, et al.: Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 601 (1-2): 171-8, 2006.
- Vogelstein B, Knizler K: Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 211-37.
- Kraemer KH, Slor H: Xeroderma pigmentosum. Clin Dermatol 3 (1): 33-69, 1985 Jan-Mar.
- Moriwaki S, Kraemer KH: Xeroderma pigmentosum--bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed 17 (2): 47-54, 2001.
- Ben Rekaya M, Laroussi N, Messaoud O, et al.: A founder large deletion mutation in Xeroderma pigmentosum-Variant form in Tunisia: implication for molecular diagnosis and therapy. Biomed Res Int 2014: 256245, 2014.
- Fassihi H, Sethi M, Fawcett H, et al.: Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A 113 (9): E1236-45, 2016.
- Amr K, Messaoud O, El Darouti M, et al.: Mutational spectrum of Xeroderma pigmentosum group A in Egyptian patients. Gene 533 (1): 52-6, 2014.
- Schriver C, Cleaver J, et al., eds.: Xeroderma pigmentosum and cockayne syndrome. In: Cleaver J, Kraemer K, eds.: The Metabolic and Molecular Bases of Inherited Disease. 7th ed. McGraw-Hill Book Co, 1995, pp 4397.
- Lambert WC, Gagna CE, Lambert MW: Xeroderma pigmentosum: its overlap with trichothiodystrophy, Cockayne syndrome and other progeroid syndromes. Adv Exp Med Biol 637: 128-37, 2008.
- Robbins JH, Kraemer KH, Lutzner MA, et al.: Xeroderma pigmentosum. An inherited diseases with sun sensitivity, multiple cutaneous neoplasms, and abnormal DNA repair. Ann Intern Med 80 (2): 221-48, 1974.
- Weeda G, Eveno E, Donker I, et al.: A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet 60 (2): 320-9, 1997.
- Lehmann AR: The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev 15 (1): 15-23, 2001.
- Broughton BC, Berneburg M, Fawcett H, et al.: Two individuals with features of both xeroderma pigmentosum and trichothiodystrophy highlight the complexity of the clinical outcomes of mutations in the XPD gene. Hum Mol Genet 10 (22): 2539-47, 2001.
- Goudie DR, Yuille MA, Leversha MA, et al.: Multiple self-healing squamous epitheliomata (ESS1) mapped to chromosome 9q22-q31 in families with common ancestry. Nat Genet 3 (2): 165-9, 1993.
- Goudie DR, D'Alessandro M, Merriman B, et al.: Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet 43 (4): 365-9, 2011.
- Bose S, Morgan LJ, Booth DR, et al.: The elusive multiple self-healing squamous epithelioma (MSSE) gene: further mapping, analysis of candidates, and loss of heterozygosity. Oncogene 25 (5): 806-12, 2006.
- Mabula JB, Chalya PL, Mchembe MD, et al.: Skin cancers among Albinos at a University teaching hospital in Northwestern Tanzania: a retrospective review of 64 cases. BMC Dermatol 12: 5, 2012.
- Luande J, Henschke CI, Mohammed N: The Tanzanian human albino skin. Natural history. Cancer 55 (8): 1823-8, 1985.
- Knöpfel N, Martín-Santiago A, Del Pozo LJ, et al.: Amelanotic naevoid melanoma in a 16-month-old albino infant. Clin Exp Dermatol 42 (1): 84-88, 2017.
- Iversen U, Iversen OH: Tumours of the skin. In: Templeton AC, ed.: Tumours in a Tropical Country: A Survey of Uganda, 1964-1968. Springer, 1973, pp 180-99.
- Hutton SM, Spritz RA: A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest Ophthalmol Vis Sci 49 (3): 868-72, 2008.
- Laetitia MM, Veronique K, Mamy NZ, et al.: Molecular genetic characterization of Congolese patients with oculocutaneous albinism. Eur J Med Genet 65 (11): 104611, 2022.
- Brilliant MH: The mouse p (pink-eyed dilution) and human P genes, oculocutaneous albinism type 2 (OCA2), and melanosomal pH. Pigment Cell Res 14 (2): 86-93, 2001.
- Sviderskaya EV, Bennett DC, Ho L, et al.: Complementation of hypopigmentation in p-mutant (pink-eyed dilution) mouse melanocytes by normal human P cDNA, and defective complementation by OCA2 mutant sequences. J Invest Dermatol 108 (1): 30-4, 1997.
- Mauri L, Manfredini E, Del Longo A, et al.: Clinical evaluation and molecular screening of a large consecutive series of albino patients. J Hum Genet 62 (2): 277-290, 2017.
- Inagaki K, Suzuki T, Shimizu H, et al.: Oculocutaneous albinism type 4 is one of the most common types of albinism in Japan. Am J Hum Genet 74 (3): 466-71, 2004.
- Mauri L, Barone L, Al Oum M, et al.: SLC45A2 mutation frequency in Oculocutaneous Albinism Italian patients doesn't differ from other European studies. Gene 533 (1): 398-402, 2014.
- Simeonov DR, Wang X, Wang C, et al.: DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum Mutat 34 (6): 827-35, 2013.
- Moreno-Artero E, Morice-Picard F, Lasseaux E, et al.: Oculo-Cutaneous Albinism Type 4 (OCA4): Phenotype-Genotype Correlation. Genes (Basel) 13 (12): , 2022.
- Ibarrola-Villava M, Hu HH, Guedj M, et al.: MC1R, SLC45A2 and TYR genetic variants involved in melanoma susceptibility in southern European populations: results from a meta-analysis. Eur J Cancer 48 (14): 2183-91, 2012.
- Kausar T, Bhatti MA, Ali M, et al.: OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin Genet 84 (1): 91-3, 2013.
- Wei AH, Zang DJ, Zhang Z, et al.: Exome sequencing identifies SLC24A5 as a candidate gene for nonsyndromic oculocutaneous albinism. J Invest Dermatol 133 (7): 1834-40, 2013.
- Morice-Picard F, Lasseaux E, François S, et al.: SLC24A5 mutations are associated with non-syndromic oculocutaneous albinism. J Invest Dermatol 134 (2): 568-571, 2014.
- Grønskov K, Dooley CM, Østergaard E, et al.: Mutations in c10orf11, a melanocyte-differentiation gene, cause autosomal-recessive albinism. Am J Hum Genet 92 (3): 415-21, 2013.
- Nathan V, Johansson PA, Palmer JM, et al.: Germline variants in oculocutaneous albinism genes and predisposition to familial cutaneous melanoma. Pigment Cell Melanoma Res 32 (6): 854-863, 2019.
- Tomita Y, Miyamura Y: Oculocutaneous albinism and analysis of tyrosinase gene in Japanese patients. Nagoya J Med Sci 61 (3-4): 97-102, 1998.
- Liu N, Kong XD, Shi HR, et al.: Tyrosinase gene mutations in the Chinese Han population with OCA1. Genet Res (Camb) 96: e14, 2014.
- FROGGATT P: Albinism in Northern Ireland. Ann Hum Genet 24: 213-38, 1960.
- McLeod R, Lowry RB: Incidence of albinism in British Columbia (B.C.). Separation by hairbulb test. Clin Genet 9 (1): 77-80, 1976.
- Martínez-Frías ML, Bermejo E: Prevalence of congenital anomaly syndromes in a Spanish gypsy population. J Med Genet 29 (7): 483-6, 1992.
- Grønskov K, Ek J, Sand A, et al.: Birth prevalence and mutation spectrum in danish patients with autosomal recessive albinism. Invest Ophthalmol Vis Sci 50 (3): 1058-64, 2009.
- Chiang PW, Drautz JM, Tsai AC, et al.: A new hypothesis of OCA1B. Am J Med Genet A 146A (22): 2968-70, 2008.
- Okoro AN: Albinism in Nigeria. A clinical and social study. Br J Dermatol 92 (5): 485-92, 1975.
- Kagore F, Lund PM: Oculocutaneous albinism among schoolchildren in Harare, Zimbabwe. J Med Genet 32 (11): 859-61, 1995.
- Lee ST, Nicholls RD, Bundey S, et al.: Mutations of the P gene in oculocutaneous albinism, ocular albinism, and Prader-Willi syndrome plus albinism. N Engl J Med 330 (8): 529-34, 1994.
- WOOLF CM: ALBINISM AMONG INDIANS IN ARIZONA AND NEW MEXICO. Am J Hum Genet 17: 23-35, 1965.
- Manga P, Kromberg JG, Box NF, et al.: Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet 61 (5): 1095-101, 1997.
- Rundshagen U, Zühlke C, Opitz S, et al.: Mutations in the MATP gene in five German patients affected by oculocutaneous albinism type 4. Hum Mutat 23 (2): 106-10, 2004.
- Bertolotti A, Lasseaux E, Plaisant C, et al.: Identification of a homozygous mutation of SLC24A5 (OCA6) in two patients with oculocutaneous albinism from French Guiana. Pigment Cell Melanoma Res 29 (1): 104-6, 2016.
- Mondal M, Sengupta M, Samanta S, et al.: Molecular basis of albinism in India: evaluation of seven potential candidate genes and some new findings. Gene 511 (2): 470-4, 2012.
- Perry PK, Silverberg NB: Cutaneous malignancy in albinism. Cutis 67 (5): 427-30, 2001.
- Fukai K, Oh J, Frenk E, et al.: Linkage disequilibrium mapping of the gene for Hermansky-Pudlak syndrome to chromosome 10q23.1-q23.3. Hum Mol Genet 4 (9): 1665-9, 1995.
- Wildenberg SC, Oetting WS, Almodóvar C, et al.: A gene causing Hermansky-Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am J Hum Genet 57 (4): 755-65, 1995.
- Anikster Y, Huizing M, White J, et al.: Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat Genet 28 (4): 376-80, 2001.
- Suzuki T, Li W, Zhang Q, et al.: Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet 30 (3): 321-4, 2002.
- Zhang Q, Zhao B, Li W, et al.: Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet 33 (2): 145-53, 2003.
- Li W, Zhang Q, Oiso N, et al.: Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 35 (1): 84-9, 2003.
- Morgan NV, Pasha S, Johnson CA, et al.: A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8). Am J Hum Genet 78 (1): 160-6, 2006.
- Cullinane AR, Curry JA, Carmona-Rivera C, et al.: A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak Syndrome type 9. Am J Hum Genet 88 (6): 778-87, 2011.
- Toro J, Turner M, Gahl WA: Dermatologic manifestations of Hermansky-Pudlak syndrome in patients with and without a 16-base pair duplication in the HPS1 gene. Arch Dermatol 135 (7): 774-80, 1999.
- Dell'Angelica EC, Shotelersuk V, Aguilar RC, et al.: Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 3 (1): 11-21, 1999.
- Nagle DL, Karim MA, Woolf EA, et al.: Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet 14 (3): 307-11, 1996.
- Perou CM, Moore KJ, Nagle DL, et al.: Identification of the murine beige gene by YAC complementation and positional cloning. Nat Genet 13 (3): 303-8, 1996.
- Barbosa MD, Nguyen QA, Tchernev VT, et al.: Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature 382 (6588): 262-5, 1996.
- Engle LJ, Kennett RH: Cloning, analysis, and chromosomal localization of myoxin (MYH12), the human homologue to the mouse dilute gene. Genomics 19 (3): 407-16, 1994.
- Ménasché G, Pastural E, Feldmann J, et al.: Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nat Genet 25 (2): 173-6, 2000.
- Ménasché G, Ho CH, Sanal O, et al.: Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest 112 (3): 450-6, 2003.
- Baardman R, Yenamandra VK, Duipmans JC, et al.: Novel insights into the epidemiology of epidermolysis bullosa (EB) from the Dutch EB Registry: EB more common than previously assumed? J Eur Acad Dermatol Venereol 35 (4): 995-1006, 2021.
- Has C, Hess M, Anemüller W, et al.: Epidemiology of inherited epidermolysis bullosa in Germany. J Eur Acad Dermatol Venereol 37 (2): 402-410, 2023.
- Petrof G, Papanikolaou M, Martinez AE, et al.: The epidemiology of epidermolysis bullosa in England and Wales: data from the national epidermolysis bullosa database. Br J Dermatol 186 (5): 843-848, 2022.
- Mariath LM, Santin JT, Frantz JA, et al.: Genotype-phenotype correlations on epidermolysis bullosa with congenital absence of skin: A comprehensive review. Clin Genet 99 (1): 29-41, 2021.
- Montaudié H, Chiaverini C, Sbidian E, et al.: Inherited epidermolysis bullosa and squamous cell carcinoma: a systematic review of 117 cases. Orphanet J Rare Dis 11 (1): 117, 2016.
- Phillips GS, Huang A, Augsburger BD, et al.: A retrospective analysis of diagnostic testing in a large North American cohort of patients with epidermolysis bullosa. J Am Acad Dermatol 86 (5): 1063-1071, 2022.
- Rossi S, Castiglia D, Pisaneschi E, et al.: Immunofluorescence mapping, electron microscopy and genetics in the diagnosis and sub-classification of inherited epidermolysis bullosa: a single-centre retrospective comparative study of 87 cases with long-term follow-up. J Eur Acad Dermatol Venereol 35 (4): 1007-1016, 2021.
- Has C, Bauer JW, Bodemer C, et al.: Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol 183 (4): 614-627, 2020.
- Bruckner-Tuderman L: Hereditary skin diseases of anchoring fibrils. J Dermatol Sci 20 (2): 122-33, 1999.
- van den Akker PC, Jonkman MF, Rengaw T, et al.: The international dystrophic epidermolysis bullosa patient registry: an online database of dystrophic epidermolysis bullosa patients and their COL7A1 mutations. Hum Mutat 32 (10): 1100-7, 2011.
- Fine JD, Bruckner-Tuderman L, Eady RA, et al.: Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol 70 (6): 1103-26, 2014.
- Fine JD: Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatol 152 (11): 1231-1238, 2016.
- Fine J, Johnson L, Suchindran C, et al.: Cancer and inherited epidermolysis bullosa. In: Fine J, Bauer E, McGuire J, et al., eds.: Epidermolysis Bullosa; Clinical, Epidemiologic, and Laboratory Advances and the Findings of the National Epidermolysis Bullosa Registry. The Johns Hopkins University Press, 1999, pp 175-92.
- Fine JD, Johnson LB, Weiner M, et al.: Chemoprevention of squamous cell carcinoma in recessive dystrophic epidermolysis bullosa: results of a phase 1 trial of systemic isotretinoin. J Am Acad Dermatol 50 (4): 563-71, 2004.
- Fine JD, Johnson LB, Weiner M, et al.: Cause-specific risks of childhood death in inherited epidermolysis bullosa. J Pediatr 152 (2): 276-80, 2008.
- van den Akker PC, van Essen AJ, Kraak MM, et al.: Long-term follow-up of patients with recessive dystrophic epidermolysis bullosa in the Netherlands: expansion of the mutation database and unusual phenotype-genotype correlations. J Dermatol Sci 56 (1): 9-18, 2009.
- Feinstein JA, Jambal P, Peoples K, et al.: Assessment of the Timing of Milestone Clinical Events in Patients With Epidermolysis Bullosa From North America. JAMA Dermatol 155 (2): 196-203, 2019.
- Farhi D: Surgical management of epidermolysis bullosa: the importance of a multidisciplinary management. Int J Dermatol 46 (8): 815-6, 2007.
- Wessagowit V, Ashton GH, Mohammedi R, et al.: Three cases of de novo dominant dystrophic epidermolysis bullosa associated with the mutation G2043R in COL7A1. Clin Exp Dermatol 26 (1): 97-9, 2001.
- Cserhalmi-Friedman PB, Garzon MC, Guzman E, et al.: Maternal germline mosaicism in dominant dystrophic epidermolysis bullosa. J Invest Dermatol 117 (5): 1327-8, 2001.
- Cuadrado-Corrales N, Sánchez-Jimeno C, García M, et al.: A prevalent mutation with founder effect in Spanish Recessive Dystrophic Epidermolysis Bullosa families. BMC Med Genet 11: 139, 2010.
- Ben Brick AS, Laroussi N, Mesrati H, et al.: Mutational founder effect in recessive dystrophic epidermolysis bullosa families from Southern Tunisia. Arch Dermatol Res 306 (4): 405-11, 2014.
- Ortiz-Urda S, Garcia J, Green CL, et al.: Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science 307 (5716): 1773-6, 2005.
- Fine JD: Inherited epidermolysis bullosa. Orphanet J Rare Dis 5: 12, 2010.
- Aumailley M, Bruckner-Tuderman L, Carter WG, et al.: A simplified laminin nomenclature. Matrix Biol 24 (5): 326-32, 2005.
- Nakano A, Chao SC, Pulkkinen L, et al.: Laminin 5 mutations in junctional epidermolysis bullosa: molecular basis of Herlitz vs. non-Herlitz phenotypes. Hum Genet 110 (1): 41-51, 2002.
- Schumann H, Hammami-Hauasli N, Pulkkinen L, et al.: Three novel homozygous point mutations and a new polymorphism in the COL17A1 gene: relation to biological and clinical phenotypes of junctional epidermolysis bullosa. Am J Hum Genet 60 (6): 1344-53, 1997.
- Fine JD, Mellerio JE: Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol 61 (3): 367-84; quiz 385-6, 2009.
- Kiritsi D, Kern JS, Schumann H, et al.: Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J Med Genet 48 (7): 450-7, 2011.
- Majewski S, Jabłońska S: Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Arch Dermatol 131 (11): 1312-8, 1995.
- Sterling JC: Human papillomaviruses and skin cancer. J Clin Virol 32 (Suppl 1): S67-71, 2005.
- Karagas MR, Nelson HH, Sehr P, et al.: Human papillomavirus infection and incidence of squamous cell and basal cell carcinomas of the skin. J Natl Cancer Inst 98 (6): 389-95, 2006.
- Ramoz N, Rueda LA, Bouadjar B, et al.: Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet 32 (4): 579-81, 2002.
- Mulvihill J, Miller R, Fraumeni J, eds.: Nosology among the neoplastic genedermatoses. In: Mulvihill J, Miller R, Fraumeni J, eds.: Genetics of Human Cancer. Raven Press, 1977, pp 145-67.
- Jabłońska S, Orth G, Jarzabek-Chorzelska M, et al.: Twenty-one years of follow-up studies of familial epidermodysplasia verruciformis. Dermatologica 158 (5): 309-27, 1979.
- McDermott DF, Gammon B, Snijders PJ, et al.: Autosomal dominant epidermodysplasia verruciformis lacking a known EVER1 or EVER2 mutation. Pediatr Dermatol 26 (3): 306-10, 2009 May-Jun.
- Lazarczyk M, Pons C, Mendoza JA, et al.: Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med 205 (1): 35-42, 2008.
- Patel AS, Karagas MR, Pawlita M, et al.: Cutaneous human papillomavirus infection, the EVER2 gene and incidence of squamous cell carcinoma: a case-control study. Int J Cancer 122 (10): 2377-9, 2008.
- Crequer A, Picard C, Patin E, et al.: Inherited MST1 deficiency underlies susceptibility to EV-HPV infections. PLoS One 7 (8): e44010, 2012.
- Ramoz N, Taïeb A, Rueda LA, et al.: Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J Invest Dermatol 114 (6): 1148-53, 2000.
- Rosenberg PS, Tamary H, Alter BP: How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A 155A (8): 1877-83, 2011.
- Rosenberg PS, Greene MH, Alter BP: Cancer incidence in persons with Fanconi anemia. Blood 101 (3): 822-6, 2003.
- Alter BP: Cancer in Fanconi anemia, 1927-2001. Cancer 97 (2): 425-40, 2003.
- Puligandla B, Stass SA, Schumacher HR, et al.: Terminal deoxynucleotidyl transferase in Fanconi's anaemia. Lancet 2 (8102): 1263, 1978.
- Alter BP, Frissora CL, Halpérin DS, et al.: Fanconi's anaemia and pregnancy. Br J Haematol 77 (3): 410-8, 1991.
- Berger R, Le Coniat M, Schaison G: Chromosome abnormalities in bone marrow of Fanconi anemia patients. Cancer Genet Cytogenet 65 (1): 47-50, 1993.
- Lebbé C, Pinquier L, Rybojad M, et al.: Fanconi's anaemia associated with multicentric Bowen's disease and decreased NK cytotoxicity. Br J Dermatol 129 (5): 615-8, 1993.
- Bagby GC, Alter BP: Fanconi anemia. Semin Hematol 43 (3): 147-56, 2006.
- Chang L, Yuan W, Zeng H, et al.: Whole exome sequencing reveals concomitant mutations of multiple FA genes in individual Fanconi anemia patients. BMC Med Genomics 7: 24, 2014.
- Wang W: Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 8 (10): 735-48, 2007.
- Howlett NG, Taniguchi T, Olson S, et al.: Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297 (5581): 606-9, 2002.
- Seal S, Thompson D, Renwick A, et al.: Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet 38 (11): 1239-41, 2006.
- Berwick M, Satagopan JM, Ben-Porat L, et al.: Genetic heterogeneity among Fanconi anemia heterozygotes and risk of cancer. Cancer Res 67 (19): 9591-6, 2007.
- Rafnar T, Sigurjonsdottir GR, Stacey SN, et al.: Association of BRCA2 K3326* With Small Cell Lung Cancer and Squamous Cell Cancer of the Skin. J Natl Cancer Inst 110 (9): 967-974, 2018.
- Walne AJ, Vulliamy T, Beswick R, et al.: TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112 (9): 3594-600, 2008.
- Alter BP, Giri N, Savage SA, et al.: Cancer in dyskeratosis congenita. Blood 113 (26): 6549-57, 2009.
- Vulliamy T, Dokal I: Dyskeratosis congenita. Semin Hematol 43 (3): 157-66, 2006.
- Knight SW, Heiss NS, Vulliamy TJ, et al.: X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am J Hum Genet 65 (1): 50-8, 1999.
- Vulliamy T, Marrone A, Szydlo R, et al.: Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat Genet 36 (5): 447-9, 2004.
- Vulliamy TJ, Walne A, Baskaradas A, et al.: Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol Dis 34 (3): 257-63, 2005 May-Jun.
- Vulliamy T, Beswick R, Kirwan M, et al.: Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A 105 (23): 8073-8, 2008.
- Walne AJ, Vulliamy T, Marrone A, et al.: Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet 16 (13): 1619-29, 2007.
- Savage SA, Giri N, Baerlocher GM, et al.: TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82 (2): 501-9, 2008.
- Marrone A, Walne A, Tamary H, et al.: Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110 (13): 4198-205, 2007.
- Ballew BJ, Yeager M, Jacobs K, et al.: Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet 132 (4): 473-80, 2013.
- Walne AJ, Vulliamy T, Kirwan M, et al.: Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet 92 (3): 448-53, 2013.
- Fedick AM, Shi L, Jalas C, et al.: Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin Genet 88 (2): 177-81, 2015.
- Batista LF, Pech MF, Zhong FL, et al.: Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474 (7351): 399-402, 2011.
- Neveling K, Bechtold A, Hoehn H: Genetic instability syndromes with progeroid features. Z Gerontol Geriatr 40 (5): 339-48, 2007.
- Zhong F, Savage SA, Shkreli M, et al.: Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 25 (1): 11-6, 2011.
- Walne AJ, Vulliamy T, Beswick R, et al.: Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome. Hum Mol Genet 19 (22): 4453-61, 2010.
- Colombo EA, Bazan JF, Negri G, et al.: Novel C16orf57 mutations in patients with Poikiloderma with Neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J Rare Dis 7: 7, 2012.
- Alter BP, Baerlocher GM, Savage SA, et al.: Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 110 (5): 1439-47, 2007.
- Vulliamy TJ, Marrone A, Knight SW, et al.: Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood 107 (7): 2680-5, 2006.
- Vulliamy TJ, Dokal I: Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie 90 (1): 122-30, 2008.
- Borg MF, Olver IN, Hill MP: Rothmund-Thomson syndrome and tolerance of chemoradiotherapy. Australas Radiol 42 (3): 216-8, 1998.
- Haneke E, Gutschmidt E: Premature multiple Bowen's disease in poikiloderma congenitale with warty hyperkeratoses. Dermatologica 158 (5): 384-8, 1979.
- Wang LL, Levy ML, Lewis RA, et al.: Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet 102 (1): 11-7, 2001.
- Piquero-Casals J, Okubo AY, Nico MM: Rothmund-thomson syndrome in three siblings and development of cutaneous squamous cell carcinoma. Pediatr Dermatol 19 (4): 312-6, 2002 Jul-Aug.
- Petkovic M, Dietschy T, Freire R, et al.: The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci 118 (Pt 18): 4261-9, 2005.
- Wang LL, Gannavarapu A, Kozinetz CA, et al.: Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst 95 (9): 669-74, 2003.
- Kitao S, Lindor NM, Shiratori M, et al.: Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics 61 (3): 268-76, 1999.
- Werner SR, Prahalad AK, Yang J, et al.: RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun 345 (1): 403-9, 2006.
- Nakayama H: RecQ family helicases: roles as tumor suppressor proteins. Oncogene 21 (58): 9008-21, 2002.
- Ajeawung NF, Nguyen TTM, Lu L, et al.: Mutations in ANAPC1, Encoding a Scaffold Subunit of the Anaphase-Promoting Complex, Cause Rothmund-Thomson Syndrome Type 1. Am J Hum Genet 105 (3): 625-630, 2019.
- Sugrañes TA, Flanagan M, Thomas C, et al.: Age of first cancer diagnosis and survival in Bloom syndrome. Genet Med 24 (7): 1476-1484, 2022.
- Cunniff C, Djavid AR, Carrubba S, et al.: Health supervision for people with Bloom syndrome. Am J Med Genet A 176 (9): 1872-1881, 2018.
- Ellis NA, Groden J, Ye TZ, et al.: The Bloom's syndrome gene product is homologous to RecQ helicases. Cell 83 (4): 655-66, 1995.
- Bugreev DV, Yu X, Egelman EH, et al.: Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev 21 (23): 3085-94, 2007.
- German J, Ellis N: Bloom syndrome. In: Vogelstein B, Kinzler KW, eds.: The Genetic Basis of Human Cancer. 2nd ed. McGraw-Hill, 2002, pp 267-88.
- German J, Sanz MM, Ciocci S, et al.: Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mutat 28 (8): 743-53, 2007.
- Ellis NA, Ciocci S, Proytcheva M, et al.: The Ashkenazic Jewish Bloom syndrome mutation blmAsh is present in non-Jewish Americans of Spanish ancestry. Am J Hum Genet 63 (6): 1685-93, 1998.
- Lu X, Lane DP: Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell 75 (4): 765-78, 1993.
- Meetei AR, Sechi S, Wallisch M, et al.: A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23 (10): 3417-26, 2003.
- Yamamoto K, Imakiire A, Miyagawa N, et al.: A report of two cases of Werner's syndrome and review of the literature. J Orthop Surg (Hong Kong) 11 (2): 224-33, 2003.
- Huang S, Lee L, Hanson NB, et al.: The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat 27 (6): 558-67, 2006.
- Goto M, Miller RW, Ishikawa Y, et al.: Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5 (4): 239-46, 1996.
- Tsuchiya H, Tomita K, Ohno M, et al.: Werner's syndrome combined with quintuplicate malignant tumors: a case report and review of literature data. Jpn J Clin Oncol 21 (2): 135-42, 1991.
- Lauper JM, Krause A, Vaughan TL, et al.: Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS One 8 (4): e59709, 2013.
- Machino H, Miki Y, Teramoto T, et al.: Cytogenetic studies in a patient with porokeratosis of Mibelli, multiple cancers and a forme fruste of Werner's syndrome. Br J Dermatol 111 (5): 579-86, 1984.
- Goto M, Imamura O, Kuromitsu J, et al.: Analysis of helicase gene mutations in Japanese Werner's syndrome patients. Hum Genet 99 (2): 191-3, 1997.
- Oshima J, Yu CE, Piussan C, et al.: Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum Mol Genet 5 (12): 1909-13, 1996.
- Uhrhammer NA, Lafarge L, Dos Santos L, et al.: Werner syndrome and mutations of the WRN and LMNA genes in France. Hum Mutat 27 (7): 718-9, 2006.
- Yu CE, Oshima J, Wijsman EM, et al.: Mutations in the consensus helicase domains of the Werner syndrome gene. Werner's Syndrome Collaborative Group. Am J Hum Genet 60 (2): 330-41, 1997.
- Shen JC, Loeb LA: The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet 16 (5): 213-20, 2000.
- Shen J, Loeb LA: Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev 122 (9): 921-44, 2001.
- Brosh RM, Bohr VA: Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp Gerontol 37 (4): 491-506, 2002.
- Furuichi Y: Premature aging and predisposition to cancers caused by mutations in RecQ family helicases. Ann N Y Acad Sci 928: 121-31, 2001.
- Lebel M: Werner syndrome: genetic and molecular basis of a premature aging disorder. Cell Mol Life Sci 58 (7): 857-67, 2001.
- Bohr VA, Brosh RM, von Kobbe C, et al.: Pathways defective in the human premature aging disease Werner syndrome. Biogerontology 3 (1-2): 89-94, 2002.
- Chen L, Oshima J: Werner Syndrome. J Biomed Biotechnol 2 (2): 46-54, 2002.
- Opresko PL, Cheng WH, von Kobbe C, et al.: Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 24 (5): 791-802, 2003.
- Pirzio LM, Pichierri P, Bignami M, et al.: Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol 180 (2): 305-14, 2008.
- Crabbe L, Jauch A, Naeger CM, et al.: Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci U S A 104 (7): 2205-10, 2007.
- Friedrich K, Lee L, Leistritz DF, et al.: WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet 128 (1): 103-11, 2010.
- Matsumoto T, Imamura O, Yamabe Y, et al.: Mutation and haplotype analyses of the Werner's syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum Genet 100 (1): 123-30, 1997.
- Satoh M, Imai M, Sugimoto M, et al.: Prevalence of Werner's syndrome heterozygotes in Japan. Lancet 353 (9166): 1766, 1999.
- Moser MJ, Oshima J, Monnat RJ: WRN mutations in Werner syndrome. Hum Mutat 13 (4): 271-9, 1999.
- Goto M, Yamabe Y, Shiratori M, et al.: Immunological diagnosis of Werner syndrome by down-regulated and truncated gene products. Hum Genet 105 (4): 301-7, 1999.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Ferrucci LM, Cartmel B, Molinaro AM, et al.: Host phenotype characteristics and MC1R in relation to early-onset basal cell carcinoma. J Invest Dermatol 132 (4): 1272-9, 2012.
- Chen AC, Martin AJ, Choy B, et al.: A Phase 3 Randomized Trial of Nicotinamide for Skin-Cancer Chemoprevention. N Engl J Med 373 (17): 1618-26, 2015.
- Tamura D, DiGiovanna JJ, Khan SG, et al.: Living with xeroderma pigmentosum: comprehensive photoprotection for highly photosensitive patients. Photodermatol Photoimmunol Photomed 30 (2-3): 146-52, 2014 Apr-Jun.
- Tamura D, DiGiovanna JJ, Kraemer KH: Xeroderma pigmentosum. In: Lebwohl MG, Birth-Jones J, Heymann WR, et al., eds.: Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 3rd ed. Saunders Elsevier, 2010, pp 789-92.
- Kraemer KH, DiGiovanna JJ, Moshell AN, et al.: Prevention of skin cancer in xeroderma pigmentosum with the use of oral isotretinoin. N Engl J Med 318 (25): 1633-7, 1988.
- DiGiovanna JJ: Retinoid chemoprevention in the high-risk patient. J Am Acad Dermatol 39 (2 Pt 3): S82-5, 1998.
- DiGiovanna J: Oral isotretinoin chemoprevention of skin cancer in xeroderma pigmentosum. J Eur Acad Derm Venereol 5 (Suppl 1): 27, 1995.
- Otley CC, Stasko T, Tope WD, et al.: Chemoprevention of nonmelanoma skin cancer with systemic retinoids: practical dosing and management of adverse effects. Dermatol Surg 32 (4): 562-8, 2006.
- Mellerio JE, Robertson SJ, Bernardis C, et al.: Management of cutaneous squamous cell carcinoma in patients with epidermolysis bullosa: best clinical practice guidelines. Br J Dermatol 174 (1): 56-67, 2016.
- Yarosh D, Klein J, O'Connor A, et al.: Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet 357 (9260): 926-9, 2001.
- Boussen H, Zwik J, Mili-Boussen I, et al.: [Therapeutic results of 5-fluorouracil in multiple and unresectable facial carcinoma secondary to xeroderma pigmentosum] Therapie 56 (6): 751-4, 2001 Nov-Dec.
- Sarasin A: Progress and prospects of xeroderma pigmentosum therapy. Adv Exp Med Biol 637: 144-51, 2008.
- Paganelli A, Giordano E, Fiorentini C, et al.: Surgical management and oncological follow-up of cutaneous squamous cell carcinomas arising in epidermolysis bullosa patients. Int J Dermatol 61 (10): 1171-1174, 2022.
- Mellerio JE, Weiner M, Denyer JE, et al.: Medical management of epidermolysis bullosa: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol 46 (8): 795-800, 2007.
- Hammersen J, Neuner A, Wild F, et al.: Attenuation of Severe Generalized Junctional Epidermolysis Bullosa by Systemic Treatment with Gentamicin. Dermatology 235 (4): 315-322, 2019.
- Wagner JE, Ishida-Yamamoto A, McGrath JA, et al.: Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med 363 (7): 629-39, 2010.
- El-Darouti M, Fawzy M, Amin I, et al.: Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatol Ther 29 (2): 96-100, 2016 Mar-Apr.
- Hainzl S, Peking P, Kocher T, et al.: COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol Ther 25 (11): 2573-2584, 2017.
- Shinkuma S, Guo Z, Christiano AM: Site-specific genome editing for correction of induced pluripotent stem cells derived from dominant dystrophic epidermolysis bullosa. Proc Natl Acad Sci U S A 113 (20): 5676-81, 2016.
- Siprashvili Z, Nguyen NT, Gorell ES, et al.: Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 316 (17): 1808-1817, 2016.
- Webber BR, Osborn MJ, McElroy AN, et al.: CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen Med 1: , 2016.
- Mosallaei D, Hao M, Antaya RJ, et al.: Molecular and Clinical Outcomes After Intravenous Gentamicin Treatment for Patients With Junctional Epidermolysis Bullosa Caused by Nonsense Variants. JAMA Dermatol 158 (4): 366-374, 2022.
- Guide SV, Gonzalez ME, Bağcı IS, et al.: Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. N Engl J Med 387 (24): 2211-2219, 2022.
Melanoma
Introducción
Se han identificado factores genéticos infrecuentes de penetrancia alta y factores genéticos comunes de penetrancia baja para el melanoma; cerca del 5 % al 10 % de todos los melanomas surgen en familias con múltiples casos. Sin embargo, una fracción significativa de estas familias no tiene variantes patogénicas detectables de genes de susceptibilidad específicos. La frecuencia de estudio de las familias con múltiples casos y las variantes genéticas específicas que se identifican difiere bastante entre poblaciones y regiones geográficas. En un estudio poblacional grande se concluyó que CDKN2A, un gen de susceptibilidad de penetrancia alta, no contribuye de manera importante a la incidencia de melanoma.[1]
El melanoma también se presenta en otras partes del cuerpo, como el ojo y el encéfalo. Para obtener más información sobre el melanoma en el ojo, consultar Tratamiento del melanoma ocular uveal.
Factores de riesgo del melanoma
Esta sección se centra en los factores de riesgo en las personas con un aumento del riesgo hereditario de presentar melanoma. Para obtener más información sobre los factores de riesgo de melanoma en la población general, consultar Prevención del cáncer de piel.
Exposición solar
La exposición al sol está bien establecida como un factor etiológico importante en todos los tipos de cáncer de piel, aunque sus efectos difieren según el tipo de cáncer. La relación entre la exposición al sol, el uso de protector solar y el desarrollo de cáncer de piel es compleja. Esta relación se complica por un factor de confusión negativo (es decir, las personas que son muy sensibles al sol suelen evitar de manera deliberada las actividades que los exponen a la luz solar directa, y es más probable que usen protector solar cuando se exponen al sol). Estas personas tienen una susceptibilidad genética al desarrollo de cáncer de piel en virtud de su fenotipo cutáneo y, por lo tanto, pueden presentar cáncer de piel independientemente de la cantidad de exposición a la luz solar o el factor de protección solar del filtro solar.[2,3]
Características pigmentarias
Las características pigmentarias son determinantes importantes de la susceptibilidad al melanoma. Hay una correlación inversa entre el riesgo de melanoma y el color de la piel que va de la piel más clara a la piel más oscura. Los grupos étnicos de piel oscura tienen un riesgo muy bajo de melanoma en las superficies pigmentadas de la piel; sin embargo, estas personas pueden presentar melanoma en superficies acrales menos pigmentadas (palmas de las manos, plantas de los pies y lechos ungueales) con la misma frecuencia que quienes tienen piel clara. En las personas de piel relativamente clara, el color de la piel se modifica por la genética y el comportamiento. El gen del receptor de la melanocortina 1 (MC1R) es uno de los principales genes que controlan la pigmentación. Para obtener más información, consultar la sección MC1R en la sección Melanoma. Otros genes pigmentarios están en investigación.[4] Un área relativamente nueva de investigación es la evaluación de múltiples variantes al mismo tiempo, que a menudo se conoce como puntuación de riesgo poligénico (PRS).[5,6,7] Las PRS para el melanoma cutáneo incluyen muchas variantes genéticas con repercusión en la expresión de genes importantes para los fenotipos pigmentados.[8] La integración de estos modelos con los factores de riesgo fenotípicos y la utilidad clínica general para la predicción del riesgo de melanoma no está clara.[9] En la práctica clínica actual no se dispone de PRS para melanoma.
Desde el punto de vista clínico, se evalúan varias características pigmentarias para calcular el riesgo de melanoma y otros tipos de cáncer de piel. Entre ellos, se encuentran los siguientes:
- Tipo de piel de Fitzpatrick. Los siguientes 6 fenotipos cutáneos se definieron a partir de la respuesta de la piel a la exposición solar al comienzo del verano.[10]
- Tipo I: Piel muy clara que siempre se quema y nunca se broncea.
- Tipo II: Piel clara que siempre se quema y a veces se broncea.
- Tipo III: Piel de pigmentación intermedia que a veces se quema y siempre se broncea.
- Tipo IV: Piel trigueña que casi nunca se quema y siempre se broncea.
- Tipo V: Piel morena moderadamente pigmentada que nunca se quema y siempre se broncea.
- Tipo VI: Piel oscura muy pigmentada que nunca se quema y siempre se broncea.
- Número de nevos o densidad de nevos.
- Nevos atípicos o anormales.
- Pecas.
Nevos o nevoides
Los nevos (lunares) son lesiones pigmentadas benignas y circunscritas en la piel o mucosas que se forman por acumulación de melanocitos. Los pacientes con múltiples nevos exhiben un aumento del riesgo de melanoma. Aunque hay evidencia de que tanto la presencia de múltiples nevos como de múltiples nevos atípicos se asocia con aumento del riesgo de melanoma, en la mayoría de los estudios se observó un riesgo más alto de melanoma en presencia de nevos atípicos.[11,12,13,14] Además, los pacientes con múltiples nevos atípicos, sin importar los antecedentes personales o familiares de melanoma, exhiben un riesgo significativamente más alto de melanoma que los pacientes sin nevos atípicos.[15] En un estudio poblacional del Reino Unido sobre factores de riesgo genético para la aparición de nevos, se observó que algunas de las mismas variantes exhiben una asociación moderada con el riesgo de melanoma.[16]
El fenotipo de múltiples nevos tiene modificadores familiares y ambientales. El número de nevos aumenta con la exposición solar durante la niñez.[17,18] El análisis de esta asociación es complejo porque el uso de la protección solar exhibe una correlación sólida con la exposición al sol. La herencia de fenotipos específicos para un gran número de nevos, incluso un gran número de nevos atípicos desde el punto de vista clínico, se consideró inicialmente que era un rasgo autosómico dominante llamado síndrome de nevo displásico [19] y síndrome de melanoma familiar con lunares múltiples atípicos.[20] Una parte de su fenotipo heredado se atribuye al principal gen de riesgo de melanoma CDKN2A que se analiza más adelante. Incluso dentro de los portadores de este gen en familias de riesgo alto, la exposición solar afecta el número de nevos.[21]
Historia familiar
Por lo general, la historia familiar de melanoma aumenta el riesgo de melanoma casi al doble. En un estudio de un registro de cáncer familiar se evaluaron cerca de 20 000 personas con melanoma y se encontró una razón de incidencia estandarizada (RIE) de 2,62 para la descendencia de personas con melanoma y de 2,94 para hermanos o hermanas.[22] En un estudio grande de más de 200 000 participantes del Nurses' Health Study, el Nurses' Health Study 2 y el Health Professionals Follow-up Study, se encontró que las personas con una historia familiar de melanoma tenían un aumento del riesgo de melanoma (cociente de riesgos instantáneos [CRI], 1,74; intervalo de confianza [IC] 95 %, 1,45–2,09).[23] Se encontró un riesgo un poco más alto en un estudio poblacional de 1 506 961 personas de Australia occidental; los familiares de primer grado (FPG) de 5660 personas con melanoma exhibieron un CRI de melanoma de 3,58 (IC 95 %, 2,43–5,43).[24] En otro estudio poblacional de más de 238 000 FPG de 23 000 pacientes con melanoma se encontró un riesgo acumulado de por vida de melanoma del 2,5 % al 3 %, que es casi el doble del riesgo de la población general.[25] El riesgo que se basa en la historia familiar depende no solo del número de familiares que tienen melanoma si no del número de melanomas que tiene cada miembro de la familia afectado.[25] Por ejemplo, el riesgo familiar de melanoma aumentó 2,2 veces (IC 95 %, 2,2–2,3) en personas que tienen un FPG con 1 melanoma y hasta 16,3 veces (IC 95 %, 9,5–26,1) en quienes tienen un FPG con 5 o más melanomas.[25] Cuando se diagnóstica melanoma en 2 o más familiares menores de 30 años, el riesgo acumulado en los familiares aumenta hasta un 14 %.[26]
En un estudio sobre la heredabilidad del cáncer en 80 309 gemelos monocigóticos y 123 382 gemelos dicigóticos, se observó que los NMSC tienen una heredabilidad del 58 % (IC 95 %, 43–73%), lo que indica que casi la mitad del riesgo de este cáncer se debe a factores hereditarios.[27] En un estudio en el que se evaluó la contribución de la historia familiar al riesgo de melanoma, se observó que la fracción atribuible a la población osciló desde menos del 1 % en Europa del norte hasta el 6,4 % en Australia,[28] lo que indica que solo un pequeño porcentaje de casos de melanoma se deben a factores familiares. Sin embargo, hay casos infrecuentes de familias con un riesgo mucho más alto que presentan melanomas en varias generaciones y en varias personas. Para las personas de estas familias, la incidencia de melanoma es más alta en la piel protegida del sol que en la piel expuesta al sol.[29]
Se encontró que el gen de susceptibilidad al melanoma hereditario, CDKN2A, está alterado en cerca del 14 % al 43 % de las familias con 3 o más casos de melanoma. Hasta la fecha, no se han identificado variantes patogénicas en más de la mitad de las familias con múltiples casos de melanoma.[30,31,32,33] Se pueden considerar análisis multigénicos en familias con múltiples casos de melanoma; con este tipo de pruebas se identifican variantes patogénicas de varios genes relacionados con el melanoma. No obstante, en un estudio grande de una prueba de panel (30 genes) en familias con melanoma sin variantes patogénicas de CDKN2A y CDK4 se encontró un rendimiento diagnóstico del 4 %.[34] En un estudio italiano grande de un panel multigénico en pacientes con melanoma, se identificaron los siguientes dos factores de predicción de variantes patogénicas: 1) presencia de cáncer de páncreas y melanoma en la misma familia y 2) personas que han presentado más de 3 melanomas. Los pacientes que reciben el diagnóstico de melanoma después de los 60 años tienen menos probabilidad de presentar una variante patogénica de un gen relacionado con el melanoma.[33]
La definición de un grupo familiar de melanoma varía según la región geográfica en todo el mundo, debido a la función que desempeña la radiación ultravioleta (UV) en la patogénesis del melanoma. En regiones de gran insolación (regiones con exposición solar alta), se necesitan 3 o más miembros de la familia afectados para definir un grupo familiar afectado. En regiones con niveles más bajos de luz solar ambiental, se considera que 2 o más miembros de la familia afectados son suficientes para definir un grupo familiar. Para obtener más información sobre los motivos para considerar una prueba genética de melanoma, consulte la sección Pruebas genéticas.
Antecedentes personales de melanoma
Un melanoma anterior acarrea un riesgo alto de presentar otros melanomas primarios, en especial para las personas con los factores de riesgo más comunes de melanoma, como un fenotipo cutáneo o historia familiar compatible, una variante patogénica del gen CDKN2A, una gran cantidad de exposición al sol en la niñez y numerosos nevos atípicos. En el entorno del cáncer esporádico, cerca del 5 % de los pacientes con melanoma presentan más de un cáncer primario, mientras que en el entorno familiar la estimación correspondiente es del 30 %. Esta tasa mayor de la esperada de múltiples cánceres primarios del mismo órgano es una característica común de los síndromes de susceptibilidad hereditaria al cáncer; representa un hallazgo clínico que debería elevar el nivel de sospecha de que el melanoma de determinado paciente se relaciona con una predisposición genética al cáncer de piel subyacente. El riesgo de un melanoma primario secundario después del diagnóstico de un primer melanoma primario es de alrededor del 5 %, y es más alto en hombres y pacientes mayores.[35,36,37,38] En un estudio en Suiza de más de 65 000 personas con melanoma se encontró una RIE de 2,8 (IC 95 %, 2,3–3,4) para un segundo melanoma en personas con historia familiar de melanoma y una RIE de 2,5 (IC 95 %, 2,3–2,7) en personas sin historia familiar compatible.[39] El riesgo de un segundo melanoma aumenta cuando el primer melanoma se diagnostica antes de los 40 años (SIR, 4,7; IC 95 % 3,9–5,6 %). La RIE aumentó con el incremento del número de melanomas.
Antecedentes personales de carcinoma de células basales o de carcinoma de células escamosas
El antecedente de un carcinoma de células basales (CCB) o carcinoma de células escamosas (CCE) también se asocia con aumento del riesgo de un melanoma posterior.[40,41,42] Dependiendo del estudio, el riesgo oscila entre un aumento no significativo de melanoma con un CCE previo de 1,04 (IC 95 %, 0,13–8,18) hasta un aumento muy significativo en el riesgo de 7,94 (IC 95 %, 4,11–15,35).[43,44] Es probable que esta relación sea consecuencia de factores de riesgo compartidos (uno de ellos, exposición solar), más que de un factor de riesgo genético específico que aumenta el riesgo de ambos. Las características pigmentarias son muy importantes para la aparición de un melanoma, y el fenotipo cutáneo (descrito antes) en combinación con una exposición excesiva al sol, se relaciona con un aumento del riesgo de los tres tipos de cáncer de piel.
Principales genes asociados con el melanoma
CDKN2A/p16yp14/ARF
El principal gen relacionado con el melanoma es CDKN2A/p16, el inhibidor de la cinasa 2A dependiente de ciclina, que se encuentra en el cromosoma 9p21. Este gen tiene varios nombres (MTS1, INK4 y MLM) y es frecuente que se conozca por el nombre de la proteína que codifica, p16. Es un regulador ascendente de la vía del gen del retinoblastoma, que actúa a través del complejo formado por la ciclina D1 y la cinasa dependiente de ciclina 4. Este gen supresor de tumores se ha estudiado de manera intensiva en familias con múltiples casos de melanoma y en series poblacionales de este tipo de cáncer. CDKN2A controla el avance de las células por el ciclo celular y proporciona un mecanismo de control para mantener las células dañadas en el punto de control G1/S de manera que se haga la reparación del daño del DNA antes de la replicación celular. La pérdida de la función de los genes supresores de tumores, como el CDKN2A, es un paso crítico en la carcinogénesis de muchos sistemas tumorales.
CDKN2A codifica dos proteínas, p16INK4a y p14ARF, con función inhibitoria de la senescencia celular. La proteína que se produce cuando se transcribe el marco de lectura alternativo (ARF) para el exón 1, en lugar del marco de lectura estándar, ejerce sus efectos biológicos en la vía p53. Su efecto es de interrupción del ciclo celular en los puestos de control G1 y G2/M, complementando el bloqueo de la progresión de G1/S de p16, lo que facilita la reparación celular del daño en el DNA.
Las variantes patogénicas de CDKN2A explican entre el 35 % y el 40 % de los melanomas familiares [30] y menos del 1 % de los casos no seleccionados de melanoma.[45] En un estudio de más de 1000 personas en España se observó que el 6,6 % de las personas con melanoma tienen una historia familiar de 2 o más FPG con melanoma, y hasta el 15 % tiene una historia familiar de melanoma familiar, que incluye diagnóstico de melanoma o cáncer de páncreas en familiares de segundo grado (FSG).[46] En una serie de casos grande del Reino Unido, se describió que las variantes patogénicas de CDKN2A se encontraban en el 100 % de las familias con 7 a 10 personas afectadas con melanoma, en el 60 % al 71 % de las familias con 4 a 6 casos y en el 14 % de las familias con 2 casos.[31] En un estudio parecido que se hizo en Grecia de personas afectadas por melanoma, se encontraron variantes patogénicas de CDKN2A en el 3,3 % de los casos unitarios de melanoma, en el 22 % de los casos de melanoma familiar y en el 57 % de las personas con melanomas primarios múltiples (MPM).[47] En un estudio de 92 casos secuenciales de personas italianas con síndrome de melanoma familiar con lunares múltiples atípicos (definido como 3 o más familiares con melanoma cutáneo primario o una persona con MPM) se encontraron variantes patogénicas de CDKN2A en el 20 % de las personas, entre ellas, 3 personas no emparentadas con una variante p.D84V.[48] Con pruebas genéticas en cascada se identificaron 14 de 40 familiares no afectados de la misma familia que se sometieron a pruebas y se encontró que eran portadores de la variante patogénica de CDKN2A. Sin embargo, en otro estudio de 106 casos de melanoma familiar (definido como por lo menos 2 casos de melanoma) solo se encontraron variantes patogénicas de CDKN2A en el 8,3 % de los casos.[49] La frecuencia de variantes patogénicas de CDKN2A es de hasta el 22 % en familias con 2 casos de melanoma que además tienen otras características de melanoma hereditario, como una edad menor de 50 años en el momento del diagnóstico del cáncer o una o más personas con MPM.[50] Muchas de las variantes patogénicas reportadas entre las familias se consideran variantes con efecto fundador, que son exclusivas de poblaciones y áreas geográficas específicas.[51,52,53,54,55,56,57,58]
En un estudio de 587 personas con un melanoma primario único o MPM se encontraron variantes patogénicas de CDKN2A en el 19 % de las personas con un familiar con MPM en comparación con el 4,4 % en las personas con un melanoma primario único.[59] Las variantes patogénicas de CDKN2A se encontraron en el 29,6 % de las personas con 3 o más melanomas primarios. Se encontró una frecuencia de variantes patogénicas de CDKN2A del 58 % en las personas con más de 3 melanomas primarios y una historia familiar de melanoma (indefinida). En otro estudio más pequeño, de 46 pacientes con 3 o más melanomas primarios, se encontraron variantes patogénicas de CDKN2A en el 24 % de los participantes.[60]
Según el diseño del estudio y la población de interés, la penetrancia del melanoma en presencia de variantes patogénicas de CDKN2A varía bastante. En un estudio de 80 casos múltiples de melanoma se demostró que la penetrancia varía por país, una observación que se atribuyó a diferencias grandes en la exposición solar.[61] Por ejemplo, en Australia, la penetrancia fue del 30 % a los 50 años de edad y del 91 % a los 80 años de edad; en los Estados Unidos, el penetrancia fue del 50 % a los 50 años de edad y del 76 % a los 80 años; en Europa, la penetrancia fue del 13 % a los 50 años y del 58 % a los 80 años. Por el contrario, en una comparación de familias con una variante patogénica del gen CDKN2A en el Reino Unido y Australia, se encontró el mismo riesgo acumulado de melanoma; para portadores de CDKN2A, el riesgo de presentar melanoma es independiente del grado de radiación UV ambiental.[62] En otro estudio de personas con melanoma de 8 registros poblacionales de cáncer y 1 muestra hospitalaria de personas que notificaron antecedentes familiares de melanoma, se sometió a todos los participantes a secuenciación de CDKN2A. Se calculó una penetrancia del 14 % a los 50 años y del 28 % a los 80 años de edad.[38] La explicación de estas diferencias radica en el método de identificación de las personas sometidas a la prueba, y las estimaciones de la penetrancia aumentan con el número de familiares afectados. El método de comprobación familiar en este último estudio hizo que fuera mucho menos probable que se identificaran familias de melanoma "muy cargadas". La herencia simultánea de variantes del gen MC1R también aumenta la penetrancia de CDKN2A; por lo tanto, esta variante genética, que se describe con más detalle a continuación, es al mismo tiempo un gen de susceptibilidad de penetrancia baja y un gen modificador.[63] Para obtener más información, consultar la sección MC1R en la sección Melanoma. También se evaluaron otros locus modificadores en portadores de CDKN2A; la interleucina-9 (IL9) y GSTT1 fueron los únicos locus con efectos que alcanzaron significación estadística, lo que indica que otros factores de riesgo menor quizás interactúan con los locus de mayor riesgo.[64,65]
En un estudio se notificó una tasa de incidencia de melanoma de 9,9 por 1000 años-persona en 354 FPG y 2,1 por 1000 años-persona en 391 FSG de probandos con una variante patogénica Leiden p16 (c.225-243del19) de CDKN2A (IC de 95 % 7,4–13,3 y 1,2–3,8, respectivamente). Estos datos indican una tasa de melanoma mucho más alta que la de la población general (aumento de 12,9 veces en la incidencia) para los FSG que no se han hecho pruebas y que tienen un familiar portador de una variante patogénica del gen CDKN2A.[66]
En un estudio se compararon las características clínicas de 7695 personas con melanoma (182 pacientes con una variante patogénica del gen CDKN2A y 7513 personas sin una variante patogénica del gen CDKN2A). En los resultados se encontró que las personas con una variante patogénica del gen CDKN2A fueron significativamente más jóvenes en el momento del diagnóstico de melanoma (media de edad en el momento del diagnóstico para las personas con una variante patogénica del gen CDKN2A vs personas sin una variante patogénica del gen CDKN2A, 39,0 vs. 54,3 años; P < 0,001). Las personas con una variante patogénica del gen CDKN2A también exhibieron un aumento de la probabilidad de presentar un segundo melanoma después de 5 años en comparación con el grupo de control (tasa de incidencia acumulada a 5 años para los portadores de variantes del gen CDKN2A de un 23 %; tasa de incidencia acumulada a 5 años para las personas del grupo de control de un 2,3 %).[67] En un estudio del ámbito pediátrico en pacientes con melanoma (9–19 años) de familias predispuestas a este tipo de cáncer, también se notificó un aumento significativo en la prevalencia de melanoma (6 a 28 veces) en relación con la población general. En esta serie, se diagnosticaron MPM, antes de los 20 años, a 7 de 21 pacientes (33 %) con variantes patogénicas de CDKN2A.[68] En un estudio italiano se establecieron correlaciones entre fenotipo y genotipo en 100 familias con melanoma familiar con el fin de determinar las características clínicas predictivas de variantes patogénicas de CDKN2A. Los probandos con MPM, que tenían por lo menos un melanoma de 0,4 mm o más de grosor de Breslow y más de 3 familiares afectados tuvieron una probabilidad superior al 90 % de tener una variante patogénica del gen CDKN2A; los probandos con ninguna de estas características tuvieron menos de un 1 % de probabilidad de tener un variante patogénica del gen CDKN2A. En general, la presencia de MPM fue la característica más predictiva en este estudio.[69]
Los portadores de variantes patogénicas de CDKN2A tienen melanomas parecidos a los melanomas esporádicos. En un estudio grande se compararon las características patológicas entre los portadores de CDKN2A y las personas con melanoma esporádico y se encontraron algunas diferencias significativas, con una tendencia baja de aumento de la pigmentación entre los portadores de variantes patogénicas.[70] En otro estudio con más de 670 portadores de variantes patogénicas de CDKN2A y 1258 portadores de CDKN2A de tipo normal o variantes benignas se encontró que era más probable que los participantes con variantes patogénicas recibieran un diagnóstico a una edad temprana (mediana de edad, 38 vs. 46 años) y presentaran MPM (número promedio de melanomas, 2,3 vs. 1,4).[71] En un estudio pequeño se compararon las tasas de supervivencia general (SG) de 106 portadores de variantes patogénicas de CDKN2A y 199 no portadores sin historia familiar de melanoma. Fue más probable que los portadores de CDKN2A presentaran MPM. Sin embargo, no se encontraron diferencias significativas en la SG ni en las tasas de supervivencia específica por la enfermedad entre portadores y no portadores.[72] No obstante, se identificaron 2 variantes patogénicas de CDKN2A (p.Arg112dup, p.Pro48Leu) que son factores pronósticos en pacientes con melanoma. Después de ajustar por edad, sexo y clasificación tumoral, los portadores de estas variantes patogénicas de CDKN2A exhibieron una supervivencia específica de melanoma más precaria que las personas que no eran portadoras de CDKN2A (CRI, 2,5; IC 95 %, 1,49–2,21).[73] En un estudio temprano se indicó que las variantes somáticas de NRAS se presentan a una tasa más alta en casos de melanomas diagnosticados en Suecia en familias portadoras de variantes patogénicas de CDKN2A en comparación con los melanomas esporádicos.[74] Sin embargo, en estudios posteriores se encontró que las tasas de variantes somáticas comunes (BRAF, NRAS) fueron más bajas en los melanomas de portadores de CDKN2A que en los melanomas esporádicos del grupo de control.[75,76] Varios pacientes con variantes del gen CDKN2A tenían melanomas con variantes de BRAF y NRAS simultáneas; lo que es una situación infrecuente en melanomas esporádicos.[76] En los datos de una serie de pacientes pequeña se indicó que los pacientes con melanoma y variantes patogénicas de CDKN2A a veces presentan mejores tasas de respuesta a la inmunoterapia en comparación con los no portadores; sin embargo, se necesitan más datos sobre este tema.[77]
Se han encontrado variantes patogénicas en el exón 1ß de CDKN2A (p14ARF) en una proporción pequeña de familias negativas para variantes patogénicas p16INK4a. En un estudio de 94 familias italianas con 2 o más casos de melanoma, el 3,2 % de las familias tenían variantes de p14ARF.[78] Un paciente con una translocación equilibrada entre los cromosomas 9 y 22 que presentaba p14ARF, tenía melanoma, deficiencia en la reparación del DNA y características del síndrome de DiGeorge, como sordera y malformaciones del oído interno.[79]
Existen modelos para predecir si una persona tiene una variante patogénica de CDKN2A.[80,81] No obstante, en la era actual estos modelos ya no se usan mucho en la práctica clínica debido a la amplia disponibilidad de análisis multigénicos poco costosos.
CDKN2A, fenotipos cutáneos y otros cánceres diferentes al melanoma
En un estudio del Melanoma Genetics Consortium (GenoMEL) con 1641 familiares de probandos con melanoma, se encontró que quienes exhibían una variante patogénica del gen CDKN2A tenían más probabilidad de tener nevos atípicos que los familiares no portadores de la variante de CDKN2A (oportunidad relativa [OR], 1,65; IC 95 %, 1,18–2,28).[82] En otro estudio en Suecia de personas con MPM y 2 o más casos de melanoma en familiares de primero, segundo y tercer grado, se encontraron variantes patogénicas de CDKN2A en 43 de 100 casos. Los casos de MPM familiar con variantes del gen CDKN2A, los casos de MPM familiar con CDKN2A de tipo normal y los casos de MPM no familiares exhibieron aumento del riesgo de CCE cutáneo futuro en comparación con los controles (riesgo relativo [RR], 4,8; IC 95 %, 1,5–15,1).[83]
En los resultados del estudio Genes, Environment, and Melanoma (GEM) se observó que los FPG de portadores de variantes patogénicas de CDKN2A con melanoma tienen un aumento del riesgo de cerca del 50 % de presentar otros tipos de cáncer en comparación con los FPG de otros pacientes con melanoma.[84] Los tipos de cáncer con aumento del riesgo en esta población fueron los cánceres gastrointestinales (CRI, 2,4; IC 95 %, 1,4–3,7), el cáncer de páncreas (CRI, 7,4; IC 95 %, 2,3–18,7) y el tumor de Wilms (CRI, 40,4; IC 95 %, 3,4–352,7). En un estudio español de FPG de 66 pacientes con melanoma y variantes patogénicas de CDKN2A, también se observó un aumento de la prevalencia de otros cánceres, como el cáncer de páncreas (razón de prevalencia [RP], 2,97; IC 95 %, 1,72–5,15), pulmón (RP, 3,04; IC 95 %, 1,93–4,80) y mama (PR, 2,19; IC 95 %, 1,36–3,55).[85] En un estudio grande de registro de Suecia, en el que participaron 27 familias portadores de la variante patogénica Arg112dup de CDKN2A, se observaron casos excesivos de otros tipos de cáncer diferentes al melanoma en portadores (n = 120) y en FPG (n = 275). Para los portadores de variantes patogénicas de CDKN2A, se observó un aumento del riesgo en comparación con una población de control para el cáncer de páncreas (RR, 43,8; IC 95 %, 13,8–139), tubo digestivo superior (RR, 17,1; IC 95 %, 6,3–46,5), vías respiratorias (RR, 15,6; IC 95 %, 5,4–46,0) y mama (RR 3,0; IC 95 %, 0,9–9,9), entre otros (todos los tipos de cáncer: RR, 5,0; IC 95 %, 3,7–7,3). Los RR en los FPG fueron de 20,6 (IC 95 %, 11,6–36,7) para el cáncer de páncreas, de 6,0 (IC 95 %, 2,8–13,1) para el cáncer de vías respiratorias, de 3,3 (IC 95 %, 1,5–7,6) para el cáncer de tubo digestivo superior y de 1,9 (IC 95 %, 0,9–4,0) para el cáncer de mama; el RR para todos los tipos de cáncer fue de 2,1 (IC 95 %, 1,6–2,7). Se observó un aumento inferior del riesgo de cáncer en los FSG. También se observó una asociación significativa entre el consumo de cigarrillos y el riesgo de cáncer de páncreas, vías respiratorias y tubo digestivo superior, con una OR de 9,3 (IC 95 %, 1,9–44,7) para portadores que habían fumado alguna vez en comparación con portadores que nunca habían fumado.[86] En un estudio de personas con variantes patogénicas del gen CDKN2A que afectaban los transcritos P16 y p14ARF, se encontró aumento de la incidencia de cánceres de esófago y tumores malignos de vaina de nervio periférico, aunque la incidencia notificada para ambos tumores fue muy baja.[87]
En algunos estudios se ha identificado variantes germinales patogénicas de CDKN2A en personas con sarcoma, pero el número de casos es muy pequeño como para determinar que haya una asociación de este gen con el riesgo de sarcoma.[88,89] Un paciente con características compatibles con el síndrome de Li-Fraumeni no portaba una variante patogénica del gen TP53, pero sí exhibía deleción de CDKN2A y CDKN2B.[89] En un estudio de secuenciación de exoma completo en una familia con un síndrome similar al de Li-Fraumeni, se identificó la misma variante patogénica del gen CDKN2A en 3 familiares que presentaban un sarcoma de tejido blando.[88] En una evaluación de 474 familias con melanoma que además presentaban casos de sarcoma y familias con un síndrome similar al de Li-Fraumeni con resultado negativo para la variante 190 TP53, se encontraron otros 8 casos de pacientes con sarcoma y variantes patogénicas de CDKN2A.
Cáncer de páncreas
Un subgrupo de familias portadoras de variantes patogénicas de CDKN2A también exhiben aumento del riesgo de cáncer de páncreas.[90,91] El riesgo de por vida de cáncer de páncreas en estas familias oscila entre el 11 % y el 17 %.[92] Se notificó un RR de hasta 47,8.[93] Aunque se han identificado por lo menos 18 variantes diferentes de p16, hay variantes patogénicas específicas que acarrean un riesgo especialmente elevado de cáncer de páncreas.[30,94] Las variantes patogénicas que afectan sitios de empalme o repeticiones de anquirina fueron más frecuentes en familias con cáncer de páncreas y melanoma en comparación con las familias que solo presentaban melanoma. La variante Leiden p16 es una deleción de 19 pares de bases en el exón 2 de CDKN2A y es una variante patogénica con efecto fundador que se originó en los Países Bajos. En un estudio holandés importante de 19 familias donde 86 miembros tenían melanoma también se encontraron 19 miembros con cáncer de páncreas y se calculó un riesgo acumulado de 17 % a los 75 años. En este estudio, la mediana de edad del inicio del cáncer de páncreas fue de 58 años, parecida a la mediana de edad del cáncer de páncreas esporádico.[95] Sin embargo, otros informes indican que la edad promedio del diagnóstico es 5,8 años más temprana para los portadores de variantes patogénicas que para aquellos con cáncer de páncreas esporádico.[96] Las variaciones geográficas quizás cumplan una función determinante en el riesgo de cáncer de páncreas en estas familias portadoras de variantes patogénicas conocidas. En un estudio de varios continentes sobre las características de las variantes germinales patogénicas de CDKN2A, se encontró que las familias australianas portadoras de estas variantes no presentaban un aumento del riesgo de cáncer de páncreas.[97] También se notificaron variantes del gen CDKN2A similares en familias con cáncer de páncreas y sin este tipo de cáncer;[98] por lo tanto, hay otros factores que afectan la formación del melanoma y el cáncer de páncreas. Algunas familias con variantes patogénicas de CDKN2A quizás exhiben una presentación de cáncer de páncreas específico de sitio sin otros tipos de cáncer.[99,100,101] Por el contrario, se han descrito familias con predisposición al melanoma que no exhiben la variante patogénica del gen CDKN2A y tienen aumento del riesgo de cáncer de páncreas.[95]
En una revisión de 110 familias con múltiples casos de cáncer de páncreas, se encontraron 18 con asociaciones entre cáncer de páncreas y melanoma.[102] En solo 5 de 18 familias con casos de cáncer de páncreas y melanoma se encontraron personas con nevos displásicos múltiples. En estas 18 familias se evaluó la presencia de variantes patogénicas de CDKN2A; solo se identificaron variantes en 2 de 18 familias, ninguna con fenotipo de nevos displásicos.
Síndrome de melanoma-astrocitoma
El síndrome de melanoma-astrocitoma es otro fenotipo causado por variantes patogénicas de CDKN2A. La sospecha de la existencia de este trastorno se describió por primera vez en 1993.[103] En un estudio de 904 personas con melanoma y sus familias, se encontraron 15 familias con 17 personas que tenían melanoma y varios tipos de tumores en el sistema nervioso.[104] En otro estudio se encontró una familia con múltiples melanomas y tumores de células neurales, aparentemente causados por pérdida de la función de p14ARF o alteración en la expresión de p16.[105] También se han notificado neurofibromas plexiformes en personas con variantes deletéreas de CDKN2A.[106,107,108,109]
CDK4 y CDK6
Las cinasas dependientes de ciclina cumplen funciones importantes en la progresión de las células de la fase G1 a la fase S. CDK4 y CDK6 en combinación con las cinasas asociadas a la ciclina D aceleran el funcionamiento del ciclo celular. La fosforilación de la proteína del retinoblastoma (Rb) en G1 mediada por las cinasas dependientes de ciclina libera factores de transcripción, lo que induce la expresión génica y cambios metabólicos que anteceden a la replicación del DNA; esto permite que la célula avance a través del ciclo celular. Estos genes son de importancia conceptual porque se encuentran en la misma vía de señalización que CDKN2A.
Las variantes germinales patogénicas de CDK4 son muy infrecuentes, solo se han encontrado en unas pocas familias extensas con melanoma.[110,111,112] En todas las familias descritas se encontró una sustitución del aminoácido 24, lo que indica que esta posición es un punto caliente de variantes del gen CDK4. En 3 familias con melanoma de Letonia se encontró una sustitución R24H en el mismo haplotipo, lo que indica que podría ser una variante patogénica con efecto fundador en esta población.[113] Una variante patogénica del gen CDK4 afecta la unión de p16 con la inhibición subsecuente de la funcionalidad de CDK4. La activación constitutiva germinal de CDK4, hace que CDK4 funcione como oncogén dominante. En un estudio pequeño se observó que el riesgo de melanoma en 17 familias con variantes patogénicas de CDK4, fue similar al riesgo observado en familias con variantes del gen CDKN2A.[114] Además, los melanomas que se encuentran en las familias con variantes de CDK4 exhiben tasas similares de variantes somáticas de BRAF que los casos de melanoma en personas que no presentan variantes patogénicas germinales en CDK4. Sin embargo, dado que las variantes germinales patogénicas de CDK4 son infrecuentes, los datos son limitados.[115] A pesar de muchas similitudes, en un estudio de 54 melanomas en 15 portadores de variantes patogénicas de CDK4, 348 melanomas en 141 portadores de variantes patogénicas de CDKN2A y 157 melanomas en 104 personas no portadoras, se encontró que era más probable que los tumores de portadores de variantes del gen CDK4 surgieran en áreas diferentes al tronco (87 % versus 45 % en no portadores [P < 0,0001] y 62 % en portadores de variantes del gen CDKN2A).[116] Para obtener más información, consultar la sección CDKN2A/p16 y p14/ARF.
A pesar de que las variantes germinales de CDK6 exhiben similitud funcional a CDK4, no se han identificado en ninguna familia extensa con melanoma.[117]
Genes de mantenimiento de los telómeros
Telomerasa transcriptasa inversa (TERT)
Si bien las variantes somáticas activadoras del gen TERT suelen encontrarse en múltiples tipos de cáncer, incluso en el melanoma esporádico, las variantes patogénicas de la línea germinan del gen TERT pocas veces se identifican en familias con melanoma.[118] Una variante patogénica de la región promotora de una subunidad de TERT se encontró en una sola familia extensa alemana cuyos miembros presentaban múltiples melanomas y otros tipos de cáncer (ovario, riñón, vejiga y pulmón). Esta variante patogénica produjo aumento de la actividad promotora en ensayos de construcción.[119]. Se llevó a cabo un análisis más detallado de miembros de esta familia extensa en 2022, y en este caso se hizo énfasis en la edad temprana de diagnóstico del melanoma (mediana de edad en el momento del diagnóstico, 30 años).[120]
La frecuencia de las variantes promotoras del gen TERT en familias con melanoma no se ha investigado bien, pero en un estudio de 273 familias con 3 o más casos de melanoma solo se identificó una familia (con 7 casos de melanoma) que portaba una variante promotora c.-57 T>G en TERT.[121] En otro estudio se analizaron 202 familias españolas con 2 o más casos de melanoma, en donde no se identificó una variante patogénica germinal del gen TERT.[122]
La patogenicidad de rs2853669, una variante específica del gen TERT, todavía está por interpretarse debido a la alta prevalencia en la población general. La prevalencia estimada de esta variante en la población general es del 25 % al 29 %. En un estudio de 106 casos familiares de melanoma (definidos como por lo menos 2 casos de melanoma o MPM en el probando) se encontró que el 47 % de los casos de MPM y el 58 % de los casos de melanoma familiar portaban una variante del promotor de TERT.[49,123]
POT1
El uso de secuenciación de exoma y genoma en miembros de familias con melanoma hereditario llevó a la identificación de variantes patogénicas de cambio de sentido del gen POT1, que se manifiestan con enfermedad en muchos estudios.[124,125] Se encontró una variante patogénica de cambio de sentido Ser270Asn del gen POT1 en 5 de 56 familias no emparentadas con casos de melanoma en Italia.[124] Esta variante no se encontró en más de 2000 controles italianos. Se cree que Ser270Asn es una variante patogénica con efecto fundador, porque todas las familias con la variante exhibían el mismo haplotipo. Además, se identificaron variantes patogénicas de cambio de sentido del gen POT1, como Tyr89Cys, Arg137His y Gln623His, en otras familias con melanoma y no se observaron en controles no afectados.[124,125] En un estudio en el que se realizó la secuenciación de POT1 en casi 3000 personas con melanoma y 3300 controles, se identificaron 43 variantes de secuencia, de inmediato se predijo que 6 de ellas eran patogénicas. En estudios funcionales, se encontró otras 9 variantes de POT1 con un efecto en la función proteínica. En general, el 0,5 % de las personas con melanoma eran portadoras de una variante potencialmente patogénica en el gen POT1.[126]
En un estudio de 290 casos de melanoma familiar (definidos como familias con al menos 2 casos de melanoma) de Estados Unidos, Italia y España, se identificaron 16 portadores de POT1 en 10 familias. Fue más probable que los melanomas en portadores de variantes del gen POT1 exhibieran características morfológicas spitzoides (P < 0,001) que los melanomas de cohortes de portadores de variantes de CDKN2A, variantes de CDK4 o grupos de no portadores. Esto se presentó incluso en los melanomas diagnosticados a edades similares en las cohortes con portadores de variantes del gen CDK4, CDKN2A y POT1. Además, fue más probable que los melanomas en portadores de POT1 presentaran en los tumores linfocitos infiltrantes en cantidad moderada o alta (P < 0,001).[127] Se encontraron variantes patogénicas de POT1 en alrededor del 1,7 % al 13,5 % de las familias con melanoma que no tenían variantes patogénicas de CDKN2A o CDK4; por lo tanto, es posible que las variantes del gen POT1 también se asocien con el melanoma hereditario.[122,127]POT1 se une a regiones de repetición telomérica monocatenarias y se cree que ayuda al mantenimiento de la longitud de los telómeros. La mayoría de las variantes segregadas en familias se presentan en 2 dominios de unión a oligonucleótido u oligosacárido de la proteína, que es la porción de la proteína fundamental para la unión al DNA. Las personas portadoras de variantes patogénicas de POT1 exhiben telómeros más largos que los casos de melanoma sin variantes del gen POT1, lo que indica un vínculo entre la alteración en la longitud normal del telómero y el melanoma.[124,125] La utilidad clínica de las pruebas para este gen no se ha determinado.
ACDyTERF2IP
En un estudio, se usó secuenciación de última generación para evaluar a 510 familias con melanoma y determinar si tenían variantes patogénicas de genes del complejo de la shelterina, que protege los extremos de los cromosomas. En 6 familias se encontraron variantes del gen ACD, y en 4 familias se encontraron variantes del gen TERF2IP.[128] Las variantes del gen ACD se agruparon en el dominio de unión a POT1. Debido a que algunas de estas variantes no dieron lugar a proteínas truncadas, no se confirmó su importancia funcional.
Genes de reparación del DNA
Los pacientes de xeroderma pigmentoso (XP) con reparación defectuosa del DNA presentan un aumento de más de 1000 veces del riesgo de desarrollar melanoma. A estos pacientes se les diagnostica melanoma a una edad significativamente más temprana que a las personas de la población general; en promedio, el melanoma se diagnóstica a los 22 años en pacientes con XP.[129] La distribución anatómica de los melanomas en pacientes con XP es parecida a la de la población general.[130,131]
Los polimorfismos genéticos asociados a los genes de reparación del DNA se vinculan con un leve aumento en el riesgo de melanoma en la población general.[132] En un metanálisis de 8 estudios de casos y controles con más de 5000 casos y 7000 controles, se encontró que los portadores del polimorfismo Asp1104His en XPG presentan aumento del riesgo de melanoma (OR, 2,42; IC 95 %, 2,26–2,60).[133] Para obtener más información, consultar la sección Xeroderma pigmentoso en la sección Carcinoma de células escamosas.
Proteína 1 asociada a BRCA1 (BAP1)
De manera reciente, se ha implicado el gen BAP1 en casos de melanoma esporádico y hereditario.[134] En un principio se describió en una cohorte de pacientes con melanoma uveal. El gen BAP1 es un gen supresor de tumores que se encuentra inactivado en el 84 % de los pacientes con melanoma que tienen metástasis.[135] Aunque muchas de estas variantes son somáticas, en un paciente se encontró una variante germinal con cambio del marco de lectura. Después se describió un fenotipo asociado a variantes patogénicas de BAP1.[136] En 2 familias con múltiples tumores melanocíticos elevados, diferentes desde el punto de vista histológico y clínico de otras neoplasias melanocíticas, se encontraron variantes germinales patogénicas con inactivación de BAP1. Estos tumores, que se conocen como tumores melanocíticos intradérmicos atípicos con mutación en BAP1, o MBAIT, se encuentran por todo el cuerpo, por lo general, miden 5 mm y aparecen en la segunda década de vida. El diámetro de estos tumores es de 2 mm a 10 mm, y por lo general las personas afectadas (cerca del 67 % de los portadores de variantes patogénicas de BAP1) tienen entre 5 a más de 50 lesiones en la piel.[136,137] Se presentan casos de melanoma cutáneo en estas familias, pero la tasa de progresión maligna es baja debido a la relativa falta de melanomas en comparación con el número de tumores papulares. Este síndrome se conoce como síndrome de tumores BAP1 o síndrome COMMON (c utaneous and o cular m elanoma and atypical m elanocytic proliferation with o ther internal n eoplasms), y se hereda de manera autosómica dominante.[138] En otras investigaciones se ha reforzado la asociación entre el melanoma cutáneo familiar y el melanoma uveal en portadores de variantes del gen BAP1.[139,140,141,142,143] Sin embargo, las variantes germinales potencialmente patogénicas de BAP1 se presentan en un porcentaje bajo de casos de melanoma. En un estudio de secuenciación dirigida de más de 1109 casos de melanoma cutáneo no seleccionados solo se encontraron 7 variantes germinales patogénicas de cambio de sentido (<1 %).[45] En otras series de 1977 casos de melanoma y 754 controles se identificaron 22 variantes del gen BAP1 infrecuentes en los casos y 5 variantes infrecuentes en los controles; se confirmó que 3 de las variantes que solo se encontraron en los casos alteraban la función de BAP1 y se vincularon con historias familiares de otros tipos de cáncer asociados al gen BAP1.[144] Los hallazgos de estudios respaldan el vínculo entre el riesgo de melanoma y BAP1.[134,140,145] En una serie, el 18 % de las personas con una variante patogénica de BAP1 presentó melanoma.[140] Otros estudios indican que en los portadores de una variante de BAP1, la penetrancia del melanoma cutáneo oscila entre el 12 % y el 23 %, y que el diagnóstico se presenta a una edad promedio de 39 años (intervalo intercuartílico, 29–53 años).[134,145] Además, aunque todavía hay pocos datos, los pacientes con variantes germinales patogénicas de BAP1 quizás presenten un aumento del riesgo de adenocarcinoma de pulmón, mesotelioma, CCB y carcinoma de células claras de riñón.[137,139,141,142,146,147]
En otros estudios se reportaron variantes patogénicas de BAP1. En una familia con múltiples casos de melanoma, se describió la presencia de una variante patogénica de cambio de sentido de BAP1 (p.Leu570Val). Esta variante de cambio de sentido afectó el empalme y produjo una proteína truncada. En esta familia también se presentaron casos de melanoma uveal y paraganglioma.[146] Otra familia con una variante patogénica Y646X BAP1 notificó múltiples cánceres, incluso múltiples melanomas cutáneos, CCB, melanomas uveales y mesoteliomas.[148] Los autores formularon la hipótesis de que hay una interacción entre genética y ambiente, puesto que las variantes patogénicas de BAP1, la radiación UV y la exposición al amianto (asbesto) contribuyen a la elevada incidencia de cáncer en esta familia.
En un estudio de simulación de Markov se obtuvo una estimación de la supervivencia y repercusiones económicas de los exámenes a portadores de variantes patogénicas de BAP1 para detectar los cuatro tipos de cáncer que se asocian de manera más común con el síndrome de predisposición de BAP1 (cáncer renal, melanoma uveal, melanoma cutáneo y mesotelioma). Los investigadores encontraron que la vigilancia resultaba rentable con un costo cercano a $1265 por año de vida ganado. La supervivencia también mejoró con la vigilancia asociada a BAP1 (50,2 % de las muertes vinculadas con BAP1 se presentaron en el grupo que no se sometió a vigilancia, mientras que solo el 35,4 % de las muertes relacionadas con BAP1 se presentaron en el grupo sometido a vigilancia). La estimación del estudio fue que solo 171 muertes entre 10 000 personas del grupo de vigilancia fueron a causa de melanoma cutáneo, mientras que 768 de las muertes entre 10 000 personas del grupo de control no sometido a vigilancia se debieron a melanoma cutáneo.[149]
Para obtener información sobre la detección de las variantes patogénicas de BAP1 y la vigilancia correspondiente, consultar la sección Detección sistemática y vigilancia en portadores de variantes patogénicas de BAP1.
Posibles regiones de susceptibilidad al melanoma familiar
En los estudios de genoma completo se han identificado varios locus adicionales de melanoma familiar. Se identificó un locus de susceptibilidad al melanoma en 1p22, mediante un análisis de ligamiento en 49 familias australianas que tenían al menos 3 casos de melanoma que eran negativos para variantes patogénicas de CDKN2A o CDK4.[150] Con el mapeo de deleciones de los tumores se encontró la pérdida de un pequeño intervalo de 9-Mb en la región de ligamiento más activa, pero ninguna de las familias estudiadas tenían variantes patogénicas de los genes evaluados hasta ahora.[151] En un estudio de asociación del genoma completo (GWAS) de miembros de 34 familias con melanomas de riesgo alto se encontraron 3 variantes de un solo nucleótido (SNV) en 10q25.1 vinculadas con riesgo de melanoma.[152] La OR para el riesgo de estas SNV osciló entre 6,8 a 8,4. Con el análisis de ligamiento paramétrico posterior de una familia se obtuvo una puntuación del logaritmo de las probabilidades de 1,5, mientras que las otras 2 familias evaluadas no exhibieron ligamiento. En la región genómica de interés no se identificó ningún gen candidato obvio. En 2 estudios de ligamiento de genoma completo en 35 de 42 familias de Suecia, se identificó evidencia de ligamiento en las regiones cromosómicas 3q29, 17p11-12 y 18q22.[153,154] En un estudio de ligamiento en Italia se encontraron indicios de ligamiento en el mismo locus 3q29.[155] No se ha confirmado ningún gen causal, pero se han mapeado los locus de todos los genes candidatos. Ninguno de estos locus se ha confirmado en estudios independientes.
Varios GWAS indicaron que es posible que exista un locus de riesgo de melanoma en el cromosoma 20q11 (OR, 1,27).[156,157] Esta es la ubicación del locus de ASIP, que codifica la proteína de señalización agoutí; esta proteína controla el color del pelo durante el ciclo de crecimiento del pelo en algunos mamíferos. Actúa como un antagonista de MC1R. Si bien, la variación en ASIP se asoció con diferencias en la pigmentación en humanos,[158] en los estudios iniciales no se encontró asociación entre las variantes del gen ASIP y el melanoma.[159] Sin embargo, en otros estudios se observó que la variante rs56238684 de ASIP se asocia con riesgo de MPM (OR, 2,5; IC 95 %, 1,7–3,3).[160] Además, la presencia de variantes de un factor de transcripción de ASIP, NCOA6 (que también está en el cromosoma 20), mostró una OR máxima de 1,82 para el riesgo de melanoma.[157] No obstante, no se observaron interacciones entre las variantes del gen NCOA6, las variantes del gen MC1R y el riesgo de melanoma. El mecanismo por el cual las variantes en 20q11 aumentan el riesgo de melanoma sigue sin estar claro.
Se han encontrado otros locus de riesgo en los cromosomas 2, 5, 6, 7, 9, 10, 11, 15, 16 y 22.[155,161,162,163,164,165,166] En un GWAS de melanoma publicado en el 2014 se examinaron 8 de los locus con una asociación significativa previa con el melanoma, pero sin un gen causal confirmado.[165] Los investigadores lograron confirmar 7 de 8 locus y encontraron alguna evidencia de apoyo para los 8. Entre ellos, se incluyó el locus del cromosoma 20 antes mencionado y un locus 9p21 distinto a CDKN2A. Los genes candidatos en estos locus se agrupan en conjuntos funcionales relacionados con la pigmentación cutánea y la formación de nevos, ambos rasgos asociados con el melanoma.[167] Para obtener más información sobre estos rasgos, consultar la sección Factores de riesgo del melanoma. En un metanálisis multicéntrico de 11 GWAS y 2 conjuntos de datos se incluyeron 15990 casos de melanoma cutáneo y 26409 controles. Se notificaron 5 locus de susceptibilidad al melanoma que implicaron supuestos elementos reguladores del melanocito, la biología de los telómeros y la reparación del DNA.[166]
Una base de datos pública, MelGene, mantiene listas de variantes identificadas mediante GWAS, que se han relacionado con riesgo de melanoma. MelGene también incluye una red y las potenciales relaciones funcionales entre estos genes y sus variantes.[168]
9q21 yGOLM1
Cuando se obtuvieron los primeros datos de asociación entre las variantes patogénicas de CDKN2A y el riesgo de melanoma, quedó claro que estas variantes no explicaban todos los tumores de melanoma en los que se pudo demostrar la pérdida de heterocigosidad de 9p21. De hecho, el 51 % de los casos informativos tenía deleciones que no abarcaban variantes somáticas de CDKN2A.[169] Hay datos que indican que el gen de la proteína de membrana de Golgi 1 (GOLM1), mapeada en 9q21 quizás esté involucrado con el riesgo de melanoma. La secuenciación del exoma del DNA de 12 conjuntos de primos y primas negativos para genes de riesgo de melanoma conocidos, llevó a la identificación de una variante del gen GOLM1 infrecuente (rs149739829) en 3 personas afectadas de un árbol genealógico.[170] Se identificaron dos pares adicionales de casos de melanoma relacionados con un posible alelo de riesgo. En los estudios de casos y controles familiares se observó una relación con el riesgo de melanoma (OR, 9,81; P < 0,001). En un estudio de casos y controles poblacional de 1534 casos de melanoma no seleccionados a partir de la historia familiar, y 1146 controles, hubo un aumento del riesgo de melanoma en portadores del alelo de riesgo en GOLM1 rs149739829 (OR, 2,45; P = 0,02).[170]
Genes secundarios (modificadores genéticos) en el melanoma
MC1R
El gen MC1R, también conocido como el receptor hormonal estimulante de melanocitos alfa, se encuentra en el cromosoma 8. Las variantes patogénicas de pérdida de función, de las que hay al menos 10, se asocian no solo con el pelo rojo, la piel clara y la pobre capacidad de bronceado, sino también con aumento del riesgo de cáncer de manera independiente del efecto sobre la pigmentación cutánea.[171,172,173,174] En un metanálisis integral de más de 8000 casos y 50 000 controles se observó un riesgo más alto de melanoma en personas con variantes del gen MC1R asociadas con pelo rojo; sin embargo los alelos no asociados con pelo rojo también se vinculan con aumento del riesgo de melanoma.[175] Además se encuentran otras asociaciones fenotípicas adicionales. En diferentes estudios, se encontraron variantes del gen MC1R asociadas con lentigo melanoma maligno (OR, 2,16; IC 95 %, 1,07–4,37; P = 0,044) [176] y aumento del riesgo de melanoma en personas sin pelo rojo, sin pecas, con tipo de piel de Fitzpatrick III o IV (OR resumida, 3,14; IC 95 %, 2,06–4,80).[177] En estudios agrupados de 5160 casos y 12 119 controles de 17 sitios se calculó que el riesgo de melanoma atribuible a las variantes del gen MC1R es del 28 %, lo que indica que estas variantes quizás contribuyen de manera importante al riesgo de melanoma en la población general.[177] Además, las personas con MPM tienen una probabilidad alta de portar una variante patogénica del gen MC1R. En un estudio de 46 personas con 3 o más melanomas primarios, 43 personas (93 %) presentaban una variante patogénica del gen MC1R.[60]
Se ha propuesto un sistema de puntuación para los polimorfismos de MC1R con el fin de identificar las asociaciones entre el grado de deterioro funcional en la vía de la melanogénesis, las características clínicas de los pacientes y la presentación clínica del melanoma. El sistema de clasificación inicial designó las variantes del gen MC1R con asociación fuerte con el pelo rojo y la piel clara como variantes fuertes (R) (OR, 63,3 IC 95 %, 31,9–139,6) y las variantes del gen MC1R que presentaron una asociación débil con pelo rojo y piel clara como variantes débiles (r) (OR, 5,1; IC 95 %, 2,5–11,3).[178] Este trabajo se expandió para evaluar otras variantes del gen MC1R adicionales que se valoraron de la siguiente manera: puntuación de 0 para la secuencia consenso de un alelo, puntuación de 1 para un alelo r y puntuación de 2 para un alelo R. La suma produce la puntuación final que oscila entre 0 y 4.[179] En un estudio de 1044 pacientes con melanoma, se observó que aquellos pacientes con una puntuación de 3 o más tuvieron una probabilidad más alta de presentar melanoma antes de los 50 años de edad (OR, 1,47; IC 95 %, 1,01–2,14).[180] De manera posterior se ha encontrado que la puntuación de MC1R tiene implicaciones en el beneficio de supervivencia de pacientes con melanoma. En por lo menos 3 estudios se observó un riesgo más bajo de muerte en pacientes con melanoma sin alelos MC1R con secuencia de consenso (CRI, 0,78; IC 95 %, 0,65–0,94) cuando se los comparó con aquellos que tenían al menos un alelo con secuencia de consenso de MC1R (CRI, 0,57–0,78).[179,181,182] Sin embargo, los beneficios de supervivencia asociados a los alelos R de MC1R tal vez solo se presenten en las mujeres.[182]
Las variantes del gen MC1R se asocian con aumento del riesgo de los 3 tipos de cáncer de piel. Sin embargo, cuando se añade la información del genotipo de MC1R a la información de las predicciones del riesgo de cáncer de piel (que se basan en edad, sexo y densidad de melanina cutánea) solo mejora un poco la predicción del riesgo.[183,184] Por el contrario, en un estudio en el que se examinaron predictores de melanoma de inicio temprano en estudios poblacionales y familiares, se observó que la adición de genotipos de MC1R mejoró la curva de rendimiento diagnóstico (ROC) en un 6 % en comparación con la información demográfica sola (P < 0,001). Cuando el genotipo de MC1R se combinó con el número de nevos de una persona y el antecedente de carcinoma de queratinocitos (CCB o CCE), la ROC fue de 0,78 (IC 95 %, 0,75–0,82) para los nevos autonotificados y de 0,83 (IC 95 %, 0,80–0,86) para los nevos descritos por el médico.[185]
En varios estudios se trató de cuantificar el incremento de la incidencia de melanoma en personas con variantes del gen MC1R. En un estudio de Australia con 1267 personas, se encontró que quienes presentaban un fenotipo R/R y más de 20 nevos presentaban un aumento del riesgo de melanoma de 25 veces en comparación con las personas que tenían una variante natural de MC1R y entre 0 y 4 nevos. El riesgo absoluto de melanoma (a los 75 años) fue del 23,3 % en los hombres con genotipos R/R de MC1R y más de 20 nevos. De manera similar, el riesgo absoluto de melanoma (a los 75 años) fue del 19,3% en mujeres con genotipos R/R MC1R y más de 20 nevos.[186] En un estudio de 233 jóvenes con melanoma, 932 adultos con melanoma y 932 controles, se encontró que las variantes r de MC1R eran más comunes en casos de melanoma en jóvenes (≤20 años) en comparación con los adultos de 35 años o más (OR, 1,54; IC 95 %, 1,02–2,33). La asociación fue más pronunciada en personas de 18 años o menos (OR, 1,80; IC 95 %, 1,06–3,07).[187]
En un estudio de 1791 personas se evaluó si las variantes del gen MC1R afectan el riesgo de melanoma de manera diferente en hombres y mujeres. Ser portador de 2 o más variantes del gen MC1R se asoció con aumento del riesgo tanto en mujeres (OR, 2,65; IC 95 %, 1,86–3,79; P = 0,001) como en hombres (OR, 1,65; IC 95 %, 1,14–2,38; P = 0,007). En un análisis multivariante (que incluyó otros factores de riesgo como pecas, arrugas, quemaduras solares y lentigos solares), la asociación entre el riesgo de melanoma y las variantes del gen MC1R fue significativa en las mujeres, pero no en los hombres.[188] En un estudio de 203 personas sin cáncer de piel se encontró que la variante r de MC1R (p.Val60Leu) se asoció con un número de nevos elevado en mujeres (>50 nevos; P < 0,001), pero no en varones. Sin embargo, el número de personas en el grupo de alto riesgo fue bajo.[189]
Las variantes del gen MC1R también modifican el riesgo de melanoma en personas con variantes patogénicas de CDKN2A. En un estudio de 815 portadores de variantes patogénicas de CDKN2A se buscaron 4 variantes frecuentes de MC1R no sinónimas y se encontró que tener una variante duplica el riesgo de melanoma, mientras que portar 2 o más variantes aumenta el riesgo casi 6 veces.[190] Después de la estratificación por color de pelo, el aumento del riesgo de melanoma se restringió a las personas con pelo marrón o negro. Estos datos indican que las variantes del gen MC1R aumentan el riesgo de melanoma en una manera independiente del efecto en la pigmentación. En un metanálisis de personas con variantes patogénicas de CDKN2A, se observó que aquellos con más de una variante de MC1R tenían un riesgo de melanoma casi cuatro veces más alto. Las personas con una o más variantes del gen MC1R exhibieron una disminución promedio de 10 años en la edad de inicio de 47 a 37 años.[191] Por el contrario, en un estudio de consorcio grande no se observó una disminución grande en la edad de inicio del melanoma.[190] En otro estudio de casos de melanoma y controles de Noruega, se observó que las variantes patogénicas de CDKN2A tenían un aumento del riesgo de melanoma cuando portaban las variantes Arg160Trp o Asp84Glu en MC1R (alelos R).[192]
Además de los estudios en los que se evalúa la relación entre las variantes germinales y las variantes del gen MC1R, varios grupos han evaluado si las variantes del gen MC1R se asocian con variantes somáticas de BRAF. En los estudios se indica que tal vez hay una asociación entre las variantes del gen MC1R y las mutaciones somáticas BRAF V600E.[193,194,195,196] Sin embargo, es difícil determinar la repercusión en la pigmentación de las variantes somáticas de BRAF en comparación con los efectos genéticos.
Otros genes de la pigmentación
Las variantes patogénicas de los genes del albinismo también explican una pequeña proporción de los casos de melanoma familiar. Por ejemplo, se observaron variantes del gen TYRP1, TYR y OCA2 con mayor frecuencia en un estudio de personas con melanoma cutáneo familiar en comparación con los controles poblacionales.[197] Se necesitan más estudios para confirmar estos hallazgos. Para obtener más información sobre los genes de la pigmentación que aumentan el riesgo de melanoma, consultar la sección Albinismo oculocutáneo.
MITF
Con la secuenciación del genoma completo se identificó la variante E318K del gen del factor de transcripción asociado a microftalmia (MITF) en una familia con 7 casos de melanoma.[198] MITF es un factor de transcripción que regula otros genes relacionados con el funcionamiento de los melanocitos. La variante MITF E318K altera la unión a sumo (SUMOylation) normal de MITF. Las personas con la variante MITF E318K son más propensas a tener piel clara, numerosos nevos, muchas pecas y pelo oscuro.[199,200] Las personas con la variante MITF E318K son más propensos a presentar melanomas amelanóticos en la evaluación clínica, melanomas de subtipo nodular y melanomas primarios múltiples.[160,199,200,201] Se considera que la variante MITF E318K es un alelo de riesgo moderado de melanoma, según se demostró en un metanálisis de 9 estudios publicados con datos de 331 pacientes (OR 2,37; IC 95 %, 1,89–2,97).[202] Aunque en estudios anteriores se indicó una relación entre las variantes patogénicas de MITF y el aumento de riesgo del carcinoma de células renales, en estudios más grandes no se ha confirmado esta relación.[202,203]
Otros genes de susceptibilidad al cáncer
BRCA1yBRCA2
El Breast Cancer Linkage Consortium encontró que las variantes patogénicas de BRCA2 se asociaban con aumento del riesgo de melanoma (RR, 2,58; IC 95 %, 1,3–5,2).[204] En otro estudio, se reportó que los portadores de variantes patogénicas de BRCA2 presentaban un aumento similar del riesgo de melanoma, si bien el resultado no alcanzó significación estadística.[205] Por el contrario, en un estudio grande de cohorte en los Países Bajos, se observó que el riesgo de melanoma no aumentaba en los portadores de variantes patogénicas de BRCA2; sin embargo, la incidencia esperada de melanoma fue infrecuente en esta población, este resultado refleja una diferencia de solo 2 casos de melanoma.[206] En los judíos asquenazíes con melanoma no se encontró aumento de la prevalencia de las 3 variantes patogénicas más frecuentes con efecto fundador de BRCA1 y BRCA2.[207] La evidencia vigente, no permite definir si las variantes patogénicas de BRCA1 y BRCA2 aumentan el riesgo de melanoma.[208,209] Para obtener más información en inglés sobre los genes BRCA1 y BRCA2, consultar BRCA1 and BRCA2: Cancer Risks and Management
CHEK2
Algunos estudios han evaluado otros genes de susceptibilidad al cáncer que aumentan el riesgo de melanoma. En una revisión de 6 estudios, en su mayoría, retrospectivos pequeños, sobre CHEK2, un gen que aumenta moderadamente el riesgo de cáncer de mama y otros cánceres, no se observó relación firme con el riesgo de melanoma.[210] Entre estos, en el estudio más grande, se evaluó la variante patogénica CHEK2 c.1100delC en pacientes daneses y alemanes. Este estudio incluyó a 1905 casos de melanoma y 12 860 controles. En los datos se observó que las personas con la variante patogénica CHEK2 c.1100delC tenían aumento del riesgo de melanoma (OR, 1,7; IC 95 % IC, 1,02–3,17). Los investigadores siguieron 2619 casos y 17 481 controles. En los datos se observó que las personas con la variante patogénica CHEK2 c.1100delC tenían aumento del riesgo de melanoma (OR, 1,81; IC 95 % IC, 1,07–3,05).[211] En los otros 5 estudios no se encontraron diferencias en la frecuencia de variantes patogénicas de CHEK2 entre los casos y los controles, incluyeron muy pocos casos de melanoma como para obtener conclusiones, o no evaluaron la variante patogénica CHEK2 c.1100delC por separado.[210] Para obtener más información en inglés, consultar la sección CHEK2 en Genetics of Breast and Gynecologic Cancers, y la sección CHEK2 en Genetics of Colorectal Cancer.
Síndrome de Li-Fraumeni (TP53)
En estudios longitudinales y retrospectivos de personas con cáncer de piel y síndrome de Li-Fraumeni (SLF) se encontró un riesgo acumulado del 36,3 % al 44,6 % para cualquier tipo de cáncer de piel a la edad de 70 años.[212,213] El riesgo de melanoma fue aproximadamente 7 veces más alto en las personas con SLF que en la población general, con un riesgo acumulado de melanoma del 12,6 % a la edad de 70 años (IC 95 %, 3,6–38,4 %). En cerca del 40 % de las personas con SLF y melanoma, el melanoma fue el cáncer primario, con una mediana de diagnóstico de 42 años de edad (intervalo, 21–68 años). El riesgo acumulado de CCB fue del 6,7 % a los 40 años y del 34,6 % a los 70 años (IC 95 %, 15,4–66,2 %). La incidencia de CCE fue similar a la que se observó en la población general. Para obtener más información en inglés, consultar la sección Li-Fraumeni Syndrome en Genetics of Breast and Gynecologic Cancers.
ATM
La ataxia telangiectasia (AT) es una afección genética autosómica recesiva causada por variantes patogénicas en el gen de la ataxia mutado (ATM). La proteína ATM es una cinasa parecida a fosfoinosítidos 3-cinasas que es responsable de la regulación de la proliferación de células nuevas.[214] Los pacientes con AT exhiben una amplia variedad de fenotipos, de acuerdo a la presencia de las siguientes características de la variante patogénica de ATM: 1) una variante truncadora que produce una proteína ATM inactiva, o 2) una variante que produce una proteína ATM con actividad y funcionalidad residuales.[215] La mayoría de los pacientes con AT manifiestan grados diversos de trastornos del movimiento (por ejemplo, tremor, ataxia), inmunodeficiencia y sensibilidad a la radiación ionizante o UV (por dificultades para la reparación de las rupturas del DNA bicatenario).[214] El 35 % de los pacientes con esta afección presentará cáncer antes de los 20 años de edad. Las leucemias y los linfomas son las neoplasias malignas más comunes en niños con AT.
En un estudio de cohorte grande se investigó el riesgo de cáncer en portadores de variantes del gen ATM (personas con una variante patogénica de ATM). Estas personas presentaron un riesgo bajo a moderado de melanoma (OR, 1,46; IC 95 %, 1,18–1,81).[216]
Alrededor del 0,5 % de la población general es portadora de variantes patogénicas del gen ATM.[214] Para obtener más información en inglés sobre el riesgo de cáncer y el abordaje de los portadores de variantes del gen ATM, consultar la sección ATM en Genetics of Breast and Gynecologic Cancers.
Síndrome de hamartoma tumoralPTEN(incluso el síndrome de Cowden)
El síndrome de Cowden y el síndrome de Bannayan-Riley-Ruvalcaba (BRRS) forman parte de un grupo de afecciones conocidas en conjunto como síndromes de hamartoma tumoral PTEN (PHTS). El término PHTS se refiere a cualquier paciente con una variante patogénica del gen PTEN, sin importar el cuadro clínico inicial.[217] El gen PTEN codifica una fosfatasa de especificidad doble que elimina los grupos fosfato de la tirosina, la serina y la treonina. Se publicaron criterios operacionales para el diagnóstico del síndrome de Cowden y luego se actualizaron.[218,219] Estos criterios incluyen criterios mayores, menores y patognomónicos, que abarcan manifestaciones mucocutáneas como tricolemoma y gangliocitoma displásico del cerebelo (enfermedad de Lhermitte-Duclos). Se propuso un nuevo conjunto de criterios a partir de una revisión sistemática de la bibliografía.[220] Estos criterios están en las directrices de la National Comprehensive Cancer Network (NCCN).[221] Para obtener más información en inglés sobre los síndromes de hamartoma tumoral PTEN (incluso el síndrome de Cowden), consultar Genetics of Colorectal Cancer y Genetics of Breast and Gynecologic Cancers.
El riesgo de melanoma en los portadores de PTEN es controversial. En un estudio del International Cowden Consortium (ICC) con 100 pacientes, se diagnosticó melanoma a 4 mujeres y 4 hombres, cuando solo se anticipaba menos de 1 caso de melanoma; en los resultados se observó una RIE de 28,3 para las mujeres (IC 95 %, 7,6–35,4) y de 39,4 para los hombres (IC 95 %, 10,6–100,9) (P < 0,001).[222] En este estudio del ICC, se notificó una RIE de 8,5 (IC 95 %, 4,1–15,6) en 368 portadores de variantes patogénicas de PTEN.[223] En esta cohorte, el riesgo de por vida de melanoma en portadores de variantes patogénicas de PTEN fue del 6 % (intervalo, 1,6–9,4 %). Cabe resaltar que en un estudio prospectivo posterior no se observó un riesgo de melanoma elevado en la misma población.[224] En este estudio, solo se diagnosticó melanoma a 1 de 180 portadores.
Evaluación del riesgo de melanoma
A los pacientes que tienen antecedentes personales de melanoma o nevos displásicos se les debe pedir que proporcionen información sobre la historia familiar de melanoma y otros tipos de cáncer para detectar el melanoma familiar. Es importante registrar la edad de los familiares en el momento del diagnóstico y la confirmación patológica, si se dispone de ella. La presencia de MPM en la misma persona también brinda un indicio sobre una susceptibilidad genética subyacente. Cerca del 30 % de las personas afectadas en familias extensas con melanoma hereditario tienen más de un melanoma primario, en comparación con un 4 % de los pacientes con melanoma esporádico.[225] Se debe actualizar la historia familiar de manera regular; a menudo se recomienda una revisión anual.
Hay varios modelos de predicción del riesgo de melanoma que se pueden usar en personas sin antecedentes personales de melanoma.[226] El rendimiento de los modelos difiere de acuerdo a la sensibilidad y especificidad, lo que abarca diferencias por sexo en algunos modelos. Los datos del Nurses' Health Study se usaron para crear un modelo que incluyó sexo, edad, historia familiar de melanoma, número de quemaduras solares graves, número de lunares mayores de 3 mm en las extremidades y color del cabello.[227] El estadístico de concordancia de este modelo fue de 0,62 (IC 95 %, 0,58–0,65). Otra medida del riesgo de melanoma inicial se derivó de un estudio de casos y controles en personas con melanoma o sin este en las áreas de Filadelfia y San Francisco.[228] Este modelo se enfocó en el sexo, los antecedentes de quemaduras solares con ampollas, el color de la tez, el número y tamaño de los lunares, la presencia de pecas, la presencia de daño solar en la piel, la incapacidad de broncearse, la edad y el área geográfica de los Estados Unidos. El riesgo atribuible con este modelo fue del 86 % para los hombres y del 89 % para las mujeres. Esta herramienta predictiva, la Melanoma Risk Assessment Tool, se puede consultar en Internet. Sin embargo, esta herramienta se desarrolló con una cohorte de personas, casi todos blancos, sin una historia familiar de melanoma ni NMSC. Está diseñada para que la usen profesionales de la salud, y se alienta a los pacientes a examinar los resultados durante la consulta médica. Es conveniente hacer otra validación externa antes de adoptar de manera generalizada esta herramienta en el ámbito clínico. Las organizaciones profesionales publicaron directrices para proveer una referencia para asesoramiento genético a personas con antecedentes personales de melanoma.[229] Para obtener más información, consultar la sección Historia familiar.
Pruebas genéticas
Se dispone de pruebas clínicas para identificar variantes germinales patogénicas de genes asociados con el melanoma hereditario MC1R, BAP1, BRCA2, CDK4, CDKN2A, MITF, TERT y POT1. Varios laboratorios en los Estados Unidos y en el extranjero ofrecen análisis de secuenciación del DNA de todas las regiones codificadoras de estos genes, así como análisis de deleción y duplicación. A menos que exista una variante patogénica conocida en una familia, a menudo se realizan pruebas en un panel de múltiples genes, en lugar de una secuenciación de gen único. El NCCN recomienda que se considere el asesoramiento genético y las pruebas de CDKN2A si los antecedentes personales o familiares de una persona se corresponden con una de dos opciones: 1) 3 o más melanomas cutáneos invasivos, o 2) una combinación de melanoma invasivo, cáncer de páncreas y astrocitomas. A veces también se necesitan pruebas multigénicas cuando una persona tiene antecedentes familiares de melanoma y otros cánceres, como melanoma uveal, astrocitoma, mesotelioma, cáncer de mama, cáncer de páncreas y cáncer de riñón.[230] Los expertos indican que las pruebas genéticas se obtengan después de la consulta de asesoramiento genético por profesionales certificados y con experiencia en los síndromes de cáncer de piel hereditarios.
Intervenciones
Población de alto riesgo
Atención médica a miembros de familias con predisposición al melanoma
Las personas con riesgo alto, como los familiares de primer y segundo grado en familias con predisposición al melanoma, deben recibir educación sobre la protección solar y los signos de alarma de melanoma.[66] También se recomienda el examen regular de la piel en consulta con profesionales de atención de la salud con experiencia en la evaluación de lesiones pigmentadas. En una directriz se indica el inicio del examen a los 10 años de edad, con exámenes semestrales hasta que los nevos se consideren estables, y luego exámenes anuales.[231] A estas personas se les debe enseñar métodos para que se hagan un autoexamen de la piel cada mes. Las técnicas como las fotografías de cuerpo entero y la dermoscopia ayudan a la visualización de las lesiones.[232,233] En un análisis de costo-eficacia se demostraron los beneficios de la detección sistemática para la población de riesgo alto.[234] Además, se ha observado que con vigilancia se detectan melanomas en un estadio más temprano que los melanomas diagnosticados antes de la identificación de una variante patogénica del gen CDKN2A.[235]
Las biopsias de las lesiones cutáneas en la población de alto riesgo se deben obtener con los mismos criterios que los utilizados para las lesiones en la población general. No se recomienda la extirpación preventiva de nevos sin características clínicas preocupantes. Las razones de esto son prácticas: muchas personas en estas familias tienen un gran número de nevos, y la eliminación completa de todos los nevos atípicos no es posible porque continúan apareciendo nuevos nevos. Además, puede pasar que las personas con mayor susceptibilidad al melanoma presenten un cáncer de novo, sin una lesión precursora como un nevo.[236]
Nivel de evidencia: 5
En la actualidad, la quimioprevención del melanoma en personas de alto riesgo sigue siendo un área de investigación activa; sin embargo, no se recomiendan medicamentos para la reducción del riesgo de melanoma en este momento.
Nivel de evidencia: 5
Detección sistemática y vigilancia en portadores de variantes patogénicas deBAP1
En la actualidad no hay recomendaciones estándar para la detección sistemática y el abordaje de los pacientes con variantes germinales patogénicas de BAP1. Dos grupos de expertos han emitido orientaciones sobre los protocolos para la detección sistemática del cáncer en portadores de variantes de BAP1, ambos grupos están de acuerdo en que los pacientes deben vigilarse de manera regular para detectar melanoma uveal, melanoma cutáneo y cáncer renal.[145,147] Sin embargo, estos dos grupos difieren en las recomendaciones sobre el momento de comenzar la detección y la manera de llevarla a cabo. El grupo de expertos que usó el método Delphi recomendó evaluación dermatológica anual en portadores, con examen corporal total y fotografía.[145] No hay consenso sobre el momento de inicio de la detección dermatológica. El grupo de expertos que usó el método Delphi también recomendó que los nevos clínicamente sospechosos de inactivación melanocítica de BAP1 se deben extirpar, aunque no están claros los beneficios de este procedimiento.
Nivel de evidencia: 5
Detección sistemática del cáncer de páncreas en portadores de variantes patogénicas endeCDKN2A
La detección sistemática del cáncer de páncreas en portadores de variantes patogénicas del gen CDKN2A sigue siendo un área de investigación y controversia. Actualmente, no hay métodos con eficacia comprobada para la detección sistemática del cáncer de páncreas en la población general. Sin embargo, los grupos de expertos recomiendan el uso de métodos de detección radiográfica en poblaciones específicas de riesgo alto. La NCCN recomienda que todos los pacientes con variantes patogénicas de CDKN2A se sometan a vigilancia de cáncer de páncreas con ecografía endoscópica e imágenes por resonancia magnética con colangiopancreatografía por resonancia magnética (IRM/CPRM) del abdomen, sin importar si cuentan con una historia familiar de cáncer de páncreas.[237] La NCCN recomienda comenzar la vigilancia anual del cáncer de páncreas a los 40 años o 10 años antes del primer diagnóstico de cáncer de páncreas en la familia (lo que ocurra primero). Estos métodos se suelen alternar, un año se hace la ecografía endoscópica y la IRM/CPRM el año que sigue. Se prefiere usar ambos métodos de manera asincrónica porque la IRM tiene más sensibilidad para las lesiones quísticas, mientras que la ecografía endoscópica tiene más sensibilidad para las lesiones sólidas.[238] El International Cancer of the Pancreas Screening (CAPS) Consortium revisó las directrices en el 2020 y ahora recomienda ecografía con IRM/MRCP inicial y después alternancia anual de ambos métodos.[239] Las recomendaciones del CAPS concuerdan con las recomendaciones de la NCCN en cuanto a la edad y el comienzo de la vigilancia del cáncer de páncreas. Además, el CAPS recomienda controles de la glucemia en ayunas o la hemoglobina A1c para la detección de diabetes en esta población.
Las personas con variantes patogénicas de CDKN2A son más propensas a obtener beneficio de la vigilancia del cáncer de páncreas. En un estudio se demostró que los portadores de variantes de CDKN2A con adenocarcinoma pancreático detectado por vigilancia tienen una tasa de supervivencia a 5 años del 24 % en comparación con tasas del 4 % al 7 % en las personas con adenocarcinomas pancreáticos esporádicos.[240] Sin embargo, no está claro si la aparente ventaja de supervivencia se debe a una estadificación más baja del cáncer de páncreas, a diferencias biológicas en el tumor de las personas con variantes patogénicas de CDKN2A o a otros posibles sesgos de este estudio.
Los biomarcadores séricos del cáncer de páncreas también son un área de investigación activa en esta población de riesgo alto. Sin embargo, los grupos que formulan las directrices no han aprobado el uso de biomarcadores séricos para identificar el cáncer de páncreas.
En estudios en curso se continúa investigando lo que implica una detección sistemática del cáncer de páncreas óptima para las personas con variantes patogénicas de CDKN2A y en otras personas con riesgo genético o familiar de cáncer de páncreas.
Nivel de evidencia: 3
Población general
En los siguientes resúmenes del PDQ se abordan las intervenciones de prevención, examen de detección y tratamiento para el melanoma en la población general (es decir, los pacientes que no tienen una predisposición genética al melanoma):
- Prevención del cáncer de piel
- Exámenes de detección del cáncer de piel
- Tratamiento del melanoma
Referencias:
- Berwick M, Orlow I, Hummer AJ, et al.: The prevalence of CDKN2A germ-line mutations and relative risk for cutaneous malignant melanoma: an international population-based study. Cancer Epidemiol Biomarkers Prev 15 (8): 1520-5, 2006.
- Rosso S, Zanetti R, Martinez C, et al.: The multicentre south European study 'Helios'. II: Different sun exposure patterns in the aetiology of basal cell and squamous cell carcinomas of the skin. Br J Cancer 73 (11): 1447-54, 1996.
- Berwick M: Counterpoint: sunscreen use is a safe and effective approach to skin cancer prevention. Cancer Epidemiol Biomarkers Prev 16 (10): 1923-4, 2007.
- Scherer D, Kumar R: Genetics of pigmentation in skin cancer--a review. Mutat Res 705 (2): 141-53, 2010.
- Cust AE, Drummond M, Kanetsky PA, et al.: Assessing the Incremental Contribution of Common Genomic Variants to Melanoma Risk Prediction in Two Population-Based Studies. J Invest Dermatol 138 (12): 2617-2624, 2018.
- Miles JA, Orlow I, Kanetsky PA, et al.: Relationship of Chromosome Arm 10q Variants to Occurrence of Multiple Primary Melanoma in the Population-Based Genes, Environment, and Melanoma (GEM) Study. J Invest Dermatol 139 (6): 1410-1412, 2019.
- Gu F, Chen TH, Pfeiffer RM, et al.: Combining common genetic variants and non-genetic risk factors to predict risk of cutaneous melanoma. Hum Mol Genet 27 (23): 4145-4156, 2018.
- Landi MT, Bishop DT, MacGregor S, et al.: Genome-wide association meta-analyses combining multiple risk phenotypes provide insights into the genetic architecture of cutaneous melanoma susceptibility. Nat Genet 52 (5): 494-504, 2020.
- Sordillo JE, Kraft P, Wu AC, et al.: Quantifying the Polygenic Contribution to Cutaneous Squamous Cell Carcinoma Risk. J Invest Dermatol 138 (7): 1507-1510, 2018.
- Fitzpatrick TB: The validity and practicality of sun-reactive skin types I through VI. Arch Dermatol 124 (6): 869-71, 1988.
- Roush GC, Nordlund JJ, Forget B, et al.: Independence of dysplastic nevi from total nevi in determining risk for nonfamilial melanoma. Prev Med 17 (3): 273-9, 1988.
- Halpern AC, Guerry D, Elder DE, et al.: Dysplastic nevi as risk markers of sporadic (nonfamilial) melanoma. A case-control study. Arch Dermatol 127 (7): 995-9, 1991.
- Garbe C, Büttner P, Weiss J, et al.: Risk factors for developing cutaneous melanoma and criteria for identifying persons at risk: multicenter case-control study of the Central Malignant Melanoma Registry of the German Dermatological Society. J Invest Dermatol 102 (5): 695-9, 1994.
- Tucker MA, Halpern A, Holly EA, et al.: Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA 277 (18): 1439-44, 1997.
- Marghoob AA, Kopf AW, Rigel DS, et al.: Risk of cutaneous malignant melanoma in patients with 'classic' atypical-mole syndrome. A case-control study. Arch Dermatol 130 (8): 993-8, 1994.
- Newton-Bishop JA, Chang YM, Iles MM, et al.: Melanocytic nevi, nevus genes, and melanoma risk in a large case-control study in the United Kingdom. Cancer Epidemiol Biomarkers Prev 19 (8): 2043-54, 2010.
- Azizi E, Iscovich J, Pavlotsky F, et al.: Use of sunscreen is linked with elevated naevi counts in Israeli school children and adolescents. Melanoma Res 10 (5): 491-8, 2000.
- Oliveria SA, Satagopan JM, Geller AC, et al.: Study of Nevi in Children (SONIC): baseline findings and predictors of nevus count. Am J Epidemiol 169 (1): 41-53, 2009.
- Elder DE, Goldman LI, Goldman SC, et al.: Dysplastic nevus syndrome: a phenotypic association of sporadic cutaneous melanoma. Cancer 46 (8): 1787-94, 1980.
- Lynch HT, Frichot BC, Lynch JF: Familial atypical multiple mole-melanoma syndrome. J Med Genet 15 (5): 352-6, 1978.
- Cannon-Albright LA, Meyer LJ, Goldgar DE, et al.: Penetrance and expressivity of the chromosome 9p melanoma susceptibility locus (MLM). Cancer Res 54 (23): 6041-4, 1994.
- Brandt A, Sundquist J, Hemminki K: Risk of incident and fatal melanoma in individuals with a family history of incident or fatal melanoma or any cancer. Br J Dermatol 165 (2): 342-8, 2011.
- Wei EX, Li X, Nan H: Having a first-degree relative with melanoma increases lifetime risk of melanoma, squamous cell carcinoma, and basal cell carcinoma. J Am Acad Dermatol 81 (2): 489-499, 2019.
- Ward SV, Dowty JG, Webster RJ, et al.: The aggregation of early-onset melanoma in young Western Australian families. Cancer Epidemiol 39 (3): 346-52, 2015.
- Chen T, Hemminki K, Kharazmi E, et al.: Multiple primary (even in situ) melanomas in a patient pose significant risk to family members. Eur J Cancer 50 (15): 2659-67, 2014.
- Fallah M, Pukkala E, Sundquist K, et al.: Familial melanoma by histology and age: joint data from five Nordic countries. Eur J Cancer 50 (6): 1176-83, 2014.
- Mucci LA, Hjelmborg JB, Harris JR, et al.: Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315 (1): 68-76, 2016.
- Olsen CM, Carroll HJ, Whiteman DC: Familial melanoma: a meta-analysis and estimates of attributable fraction. Cancer Epidemiol Biomarkers Prev 19 (1): 65-73, 2010.
- Hemminki K, Zhang H, Czene K: Incidence trends and familial risks in invasive and in situ cutaneous melanoma by sun-exposed body sites. Int J Cancer 104 (6): 764-71, 2003.
- Goldstein AM, Chan M, Harland M, et al.: High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res 66 (20): 9818-28, 2006.
- Bishop JN, Harland M, Bishop DT: The genetics of melanoma. Br J Hosp Med (Lond) 67 (6): 299-304, 2006.
- Pissa M, Helkkula T, Appelqvist F, et al.: CDKN2A genetic testing in melanoma-prone families in Sweden in the years 2015-2020: implications for novel national recommendations. Acta Oncol 60 (7): 888-896, 2021.
- Bruno W, Dalmasso B, Barile M, et al.: Predictors of germline status for hereditary melanoma: 5 years of multi-gene panel testing within the Italian Melanoma Intergroup. ESMO Open 7 (4): 100525, 2022.
- Potjer TP, Bollen S, Grimbergen AJEM, et al.: Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer 144 (10): 2453-2464, 2019.
- Goggins WB, Tsao H: A population-based analysis of risk factors for a second primary cutaneous melanoma among melanoma survivors. Cancer 97 (3): 639-43, 2003.
- Slingluff CL, Vollmer RT, Seigler HF: Multiple primary melanoma: incidence and risk factors in 283 patients. Surgery 113 (3): 330-9, 1993.
- Giles G, Staples M, McCredie M, et al.: Multiple primary melanomas: an analysis of cancer registry data from Victoria and New South Wales. Melanoma Res 5 (6): 433-8, 1995.
- Begg CB, Orlow I, Hummer AJ, et al.: Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst 97 (20): 1507-15, 2005.
- Chen T, Fallah M, Försti A, et al.: Risk of Next Melanoma in Patients With Familial and Sporadic Melanoma by Number of Previous Melanomas. JAMA Dermatol 151 (6): 607-15, 2015.
- Marghoob AA, Slade J, Salopek TG, et al.: Basal cell and squamous cell carcinomas are important risk factors for cutaneous malignant melanoma. Screening implications. Cancer 75 (2 Suppl): 707-14, 1995.
- Nugent Z, Demers AA, Wiseman MC, et al.: Risk of second primary cancer and death following a diagnosis of nonmelanoma skin cancer. Cancer Epidemiol Biomarkers Prev 14 (11 Pt 1): 2584-90, 2005.
- Rosenberg CA, Khandekar J, Greenland P, et al.: Cutaneous melanoma in postmenopausal women after nonmelanoma skin carcinoma: the Women's Health Initiative Observational Study. Cancer 106 (3): 654-63, 2006.
- Karagas MR, Greenberg ER, Mott LA, et al.: Occurrence of other cancers among patients with prior basal cell and squamous cell skin cancer. Cancer Epidemiol Biomarkers Prev 7 (2): 157-61, 1998.
- Chen J, Ruczinski I, Jorgensen TJ, et al.: Nonmelanoma skin cancer and risk for subsequent malignancy. J Natl Cancer Inst 100 (17): 1215-22, 2008.
- Aoude LG, Gartside M, Johansson P, et al.: Prevalence of Germline BAP1, CDKN2A, and CDK4 Mutations in an Australian Population-Based Sample of Cutaneous Melanoma Cases. Twin Res Hum Genet 18 (2): 126-33, 2015.
- Márquez-Rodas I, Martín González M, Nagore E, et al.: Frequency and characteristics of familial melanoma in Spain: the FAM-GEM-1 Study. PLoS One 10 (4): e0124239, 2015.
- Nikolaou V, Kang X, Stratigos A, et al.: Comprehensive mutational analysis of CDKN2A and CDK4 in Greek patients with cutaneous melanoma. Br J Dermatol 165 (6): 1219-22, 2011.
- Borroni RG, Manganoni AM, Grassi S, et al.: Genetic counselling and high-penetrance susceptibility gene analysis reveal the novel CDKN2A p.D84V (c.251A>T) mutation in melanoma-prone families from Italy. Melanoma Res 27 (2): 97-103, 2017.
- Pellegrini C, Maturo MG, Martorelli C, et al.: Characterization of melanoma susceptibility genes in high-risk patients from Central Italy. Melanoma Res 27 (3): 258-267, 2017.
- Maubec E, Chaudru V, Mohamdi H, et al.: Familial melanoma: clinical factors associated with germline CDKN2A mutations according to the number of patients affected by melanoma in a family. J Am Acad Dermatol 67 (6): 1257-64, 2012.
- Borg A, Johannsson U, Johannsson O, et al.: Novel germline p16 mutation in familial malignant melanoma in southern Sweden. Cancer Res 56 (11): 2497-500, 1996.
- Borg A, Sandberg T, Nilsson K, et al.: High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst 92 (15): 1260-6, 2000.
- Hashemi J, Bendahl PO, Sandberg T, et al.: Haplotype analysis and age estimation of the 113insR CDKN2A founder mutation in Swedish melanoma families. Genes Chromosomes Cancer 31 (2): 107-16, 2001.
- Ciotti P, Struewing JP, Mantelli M, et al.: A single genetic origin for the G101W CDKN2A mutation in 20 melanoma-prone families. Am J Hum Genet 67 (2): 311-9, 2000.
- Liu L, Dilworth D, Gao L, et al.: Mutation of the CDKN2A 5' UTR creates an aberrant initiation codon and predisposes to melanoma. Nat Genet 21 (1): 128-32, 1999.
- Pollock PM, Spurr N, Bishop T, et al.: Haplotype analysis of two recurrent CDKN2A mutations in 10 melanoma families: evidence for common founders and independent mutations. Hum Mutat 11 (6): 424-31, 1998.
- Larre Borges A, Borges AL, Cuéllar F, et al.: CDKN2A mutations in melanoma families from Uruguay. Br J Dermatol 161 (3): 536-41, 2009.
- de Ávila AL, Krepischi AC, Moredo LF, et al.: Germline CDKN2A mutations in Brazilian patients of hereditary cutaneous melanoma. Fam Cancer 13 (4): 645-9, 2014.
- Bruno W, Pastorino L, Ghiorzo P, et al.: Multiple primary melanomas (MPMs) and criteria for genetic assessment: MultiMEL, a multicenter study of the Italian Melanoma Intergroup. J Am Acad Dermatol 74 (2): 325-32, 2016.
- Li C, Liu T, Tavtigian SV, et al.: Targeted germline sequencing of patients with three or more primary melanomas reveals high rate of pathogenic variants. Melanoma Res 30 (3): 247-251, 2020.
- Bishop DT, Demenais F, Goldstein AM, et al.: Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst 94 (12): 894-903, 2002.
- Cust AE, Harland M, Makalic E, et al.: Melanoma risk for CDKN2A mutation carriers who are relatives of population-based case carriers in Australia and the UK. J Med Genet 48 (4): 266-72, 2011.
- Santillan AA, Cherpelis BS, Glass LF, et al.: Management of familial melanoma and nonmelanoma skin cancer syndromes. Surg Oncol Clin N Am 18 (1): 73-98, viii, 2009.
- Yang XR, Pfeiffer RM, Wheeler W, et al.: Identification of modifier genes for cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. Int J Cancer 125 (12): 2912-7, 2009.
- Chaudru V, Lo MT, Lesueur F, et al.: Protective effect of copy number polymorphism of glutathione S-transferase T1 gene on melanoma risk in presence of CDKN2A mutations, MC1R variants and host-related phenotypes. Fam Cancer 8 (4): 371-7, 2009.
- van der Rhee JI, Boonk SE, Putter H, et al.: Surveillance of second-degree relatives from melanoma families with a CDKN2A germline mutation. Cancer Epidemiol Biomarkers Prev 22 (10): 1771-7, 2013.
- van der Rhee JI, Krijnen P, Gruis NA, et al.: Clinical and histologic characteristics of malignant melanoma in families with a germline mutation in CDKN2A. J Am Acad Dermatol 65 (2): 281-8, 2011.
- Goldstein AM, Stidd KC, Yang XR, et al.: Pediatric melanoma in melanoma-prone families. Cancer 124 (18): 3715-3723, 2018.
- Pedace L, De Simone P, Castori M, et al.: Clinical features predicting identification of CDKN2A mutations in Italian patients with familial cutaneous melanoma. Cancer Epidemiol 35 (6): e116-20, 2011.
- Sargen MR, Kanetsky PA, Newton-Bishop J, et al.: Histologic features of melanoma associated with CDKN2A genotype. J Am Acad Dermatol 72 (3): 496-507.e7, 2015.
- Taylor NJ, Handorf EA, Mitra N, et al.: Phenotypic and Histopathological Tumor Characteristics According to CDKN2A Mutation Status among Affected Members of Melanoma Families. J Invest Dermatol 136 (5): 1066-9, 2016.
- Dalmasso B, Pastorino L, Ciccarese G, et al.: CDKN2A germline mutations are not associated with poor survival in an Italian cohort of melanoma patients. J Am Acad Dermatol 80 (5): 1263-1271, 2019.
- Helgadottir H, Höiom V, Tuominen R, et al.: Germline CDKN2A Mutation Status and Survival in Familial Melanoma Cases. J Natl Cancer Inst 108 (11): , 2016.
- Eskandarpour M, Hashemi J, Kanter L, et al.: Frequency of UV-inducible NRAS mutations in melanomas of patients with germline CDKN2A mutations. J Natl Cancer Inst 95 (11): 790-8, 2003.
- Zebary A, Omholt K, van Doorn R, et al.: Somatic BRAF and NRAS mutations in familial melanomas with known germline CDKN2A status: a GenoMEL study. J Invest Dermatol 134 (1): 287-290, 2014.
- Jovanovic B, Egyhazi S, Eskandarpour M, et al.: Coexisting NRAS and BRAF mutations in primary familial melanomas with specific CDKN2A germline alterations. J Invest Dermatol 130 (2): 618-20, 2010.
- Helgadottir H, Ghiorzo P, van Doorn R, et al.: Efficacy of novel immunotherapy regimens in patients with metastatic melanoma with germline CDKN2A mutations. J Med Genet 57 (5): 316-321, 2020.
- Binni F, Antigoni I, De Simone P, et al.: Novel and recurrent p14 mutations in Italian familial melanoma. Clin Genet 77 (6): 581-6, 2010.
- Tan X, Anzick SL, Khan SG, et al.: Chimeric negative regulation of p14ARF and TBX1 by a t(9;22) translocation associated with melanoma, deafness, and DNA repair deficiency. Hum Mutat 34 (9): 1250-9, 2013.
- Niendorf KB, Goggins W, Yang G, et al.: MELPREDICT: a logistic regression model to estimate CDKN2A carrier probability. J Med Genet 43 (6): 501-6, 2006.
- Wang W, Niendorf KB, Patel D, et al.: Estimating CDKN2A carrier probability and personalizing cancer risk assessments in hereditary melanoma using MelaPRO. Cancer Res 70 (2): 552-9, 2010.
- Taylor NJ, Mitra N, Goldstein AM, et al.: Germline Variation at CDKN2A and Associations with Nevus Phenotypes among Members of Melanoma Families. J Invest Dermatol 137 (12): 2606-2612, 2017.
- Helgadottir H, Tuominen R, Olsson H, et al.: Cancer risks and survival in patients with multiple primary melanomas: Association with family history of melanoma and germline CDKN2A mutation status. J Am Acad Dermatol 77 (5): 893-901, 2017.
- Mukherjee B, Delancey JO, Raskin L, et al.: Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J Natl Cancer Inst 104 (12): 953-6, 2012.
- Potrony M, Puig-Butillé JA, Aguilera P, et al.: Increased prevalence of lung, breast, and pancreatic cancers in addition to melanoma risk in families bearing the cyclin-dependent kinase inhibitor 2A mutation: implications for genetic counseling. J Am Acad Dermatol 71 (5): 888-95, 2014.
- Helgadottir H, Höiom V, Jönsson G, et al.: High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet 51 (8): 545-52, 2014.
- Sargen MR, Helgadottir H, Yang XR, et al.: Impact of Transcript (p16/p14ARF) Alteration on Cancer Risk in CDKN2A Germline Pathogenic Variant Carriers. JNCI Cancer Spectr 6 (6): , 2022.
- Jouenne F, Chauvot de Beauchene I, Bollaert E, et al.: Germline CDKN2A/P16INK4A mutations contribute to genetic determinism of sarcoma. J Med Genet 54 (9): 607-612, 2017.
- Chan SH, Lim WK, Michalski ST, et al.: Germline hemizygous deletion of CDKN2A-CDKN2B locus in a patient presenting with Li-Fraumeni syndrome. NPJ Genom Med 1: 16015, 2016.
- Goldstein AM, Fraser MC, Struewing JP, et al.: Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 333 (15): 970-4, 1995.
- Whelan AJ, Bartsch D, Goodfellow PJ: Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med 333 (15): 975-7, 1995.
- Lindor NM, McMaster ML, Lindor CJ, et al.: Concise handbook of familial cancer susceptibility syndromes - second edition. J Natl Cancer Inst Monogr (38): 1-93, 2008.
- de Snoo FA, Bishop DT, Bergman W, et al.: Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res 14 (21): 7151-7, 2008.
- Goldstein AM: Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum Mutat 23 (6): 630, 2004.
- Vasen HF, Gruis NA, Frants RR, et al.: Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer 87 (6): 809-11, 2000.
- McWilliams RR, Bamlet WR, Rabe KG, et al.: Association of family history of specific cancers with a younger age of onset of pancreatic adenocarcinoma. Clin Gastroenterol Hepatol 4 (9): 1143-7, 2006.
- Goldstein AM, Chan M, Harland M, et al.: Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet 44 (2): 99-106, 2007.
- Goldstein AM, Struewing JP, Chidambaram A, et al.: Genotype-phenotype relationships in U.S. melanoma-prone families with CDKN2A and CDK4 mutations. J Natl Cancer Inst 92 (12): 1006-10, 2000.
- Bartsch DK, Sina-Frey M, Lang S, et al.: CDKN2A germline mutations in familial pancreatic cancer. Ann Surg 236 (6): 730-7, 2002.
- Harinck F, Kluijt I, van der Stoep N, et al.: Indication for CDKN2A-mutation analysis in familial pancreatic cancer families without melanomas. J Med Genet 49 (6): 362-5, 2012.
- Zhen DB, Rabe KG, Gallinger S, et al.: BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med 17 (7): 569-77, 2015.
- Bartsch DK, Langer P, Habbe N, et al.: Clinical and genetic analysis of 18 pancreatic carcinoma/melanoma-prone families. Clin Genet 77 (4): 333-41, 2010.
- Kaufman DK, Kimmel DW, Parisi JE, et al.: A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology 43 (9): 1728-31, 1993.
- Azizi E, Friedman J, Pavlotsky F, et al.: Familial cutaneous malignant melanoma and tumors of the nervous system. A hereditary cancer syndrome. Cancer 76 (9): 1571-8, 1995.
- Randerson-Moor JA, Harland M, Williams S, et al.: A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet 10 (1): 55-62, 2001.
- Vanneste R, Smith E, Graham G: Multiple neurofibromas as the presenting feature of familial atypical multiple malignant melanoma (FAMMM) syndrome. Am J Med Genet A 161A (6): 1425-31, 2013.
- Petty EM, Bolognia JL, Bale AE, et al.: Cutaneous malignant melanoma and atypical moles associated with a constitutional rearrangement of chromosomes 5 and 9. Am J Med Genet 45 (1): 77-80, 1993.
- Petronzelli F, Sollima D, Coppola G, et al.: CDKN2A germline splicing mutation affecting both p16(ink4) and p14(arf) RNA processing in a melanoma/neurofibroma kindred. Genes Chromosomes Cancer 31 (4): 398-401, 2001.
- Bahuau M, Vidaud D, Jenkins RB, et al.: Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res 58 (11): 2298-303, 1998.
- Zuo L, Weger J, Yang Q, et al.: Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet 12 (1): 97-9, 1996.
- Soufir N, Avril MF, Chompret A, et al.: Prevalence of p16 and CDK4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma Study Group. Hum Mol Genet 7 (2): 209-16, 1998.
- Molven A, Grimstvedt MB, Steine SJ, et al.: A large Norwegian family with inherited malignant melanoma, multiple atypical nevi, and CDK4 mutation. Genes Chromosomes Cancer 44 (1): 10-8, 2005.
- Veinalde R, Ozola A, Azarjana K, et al.: Analysis of Latvian familial melanoma patients shows novel variants in the noncoding regions of CDKN2A and that the CDK4 mutation R24H is a founder mutation. Melanoma Res 23 (3): 221-6, 2013.
- Puntervoll HE, Yang XR, Vetti HH, et al.: Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet 50 (4): 264-70, 2013.
- Puntervoll HE, Molven A, Akslen LA: Frequency of somatic BRAF mutations in melanocytic lesions from patients in a CDK4 melanoma family. Pigment Cell Melanoma Res 27 (1): 149-51, 2014.
- Sargen MR, Pfeiffer RM, Yang XR, et al.: Variation in Cutaneous Patterns of Melanomagenesis According to Germline CDKN2A/CDK4 Status in Melanoma-Prone Families. J Invest Dermatol 140 (1): 174-181.e3, 2020.
- Shennan MG, Badin AC, Walsh S, et al.: Lack of germline CDK6 mutations in familial melanoma. Oncogene 19 (14): 1849-52, 2000.
- Huang FW, Hodis E, Xu MJ, et al.: Highly recurrent TERT promoter mutations in human melanoma. Science 339 (6122): 957-9, 2013.
- Horn S, Figl A, Rachakonda PS, et al.: TERT promoter mutations in familial and sporadic melanoma. Science 339 (6122): 959-61, 2013.
- Zaremba A, Meier F, Schlein C, et al.: Clinical and pathological characteristics of familial melanoma with germline TERT promoter variants. Pigment Cell Melanoma Res 35 (6): 573-586, 2022.
- Harland M, Petljak M, Robles-Espinoza CD, et al.: Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer 15 (1): 139-44, 2016.
- Potrony M, Puig-Butille JA, Ribera-Sola M, et al.: POT1 germline mutations but not TERT promoter mutations are implicated in melanoma susceptibility in a large cohort of Spanish melanoma families. Br J Dermatol 181 (1): 105-113, 2019.
- Reference SNP (refSNP) Cluster Report: rs2853669. In: Database of Short Genetic Variations (dbSNP) [Database]. Bethesda, MD: National Center for Biotechnology Information, 2018. Available online. Last accessed April 25, 2024.
- Shi J, Yang XR, Ballew B, et al.: Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet 46 (5): 482-6, 2014.
- Robles-Espinoza CD, Harland M, Ramsay AJ, et al.: POT1 loss-of-function variants predispose to familial melanoma. Nat Genet 46 (5): 478-81, 2014.
- Simonin-Wilmer I, Ossio R, Leddin EM, et al.: Population-based analysis of POT1 variants in a cutaneous melanoma case-control cohort. J Med Genet 60 (7): 692-696, 2023.
- Sargen MR, Calista D, Elder DE, et al.: Histologic features of melanoma associated with germline mutations of CDKN2A, CDK4, and POT1 in melanoma-prone families from the United States, Italy, and Spain. J Am Acad Dermatol 83 (3): 860-869, 2020.
- Aoude LG, Pritchard AL, Robles-Espinoza CD, et al.: Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst 107 (2): , 2015.
- Bradford PT, Goldstein AM, Tamura D, et al.: Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48 (3): 168-76, 2011.
- Kraemer KH, Lee MM, Scotto J: DNA repair protects against cutaneous and internal neoplasia: evidence from xeroderma pigmentosum. Carcinogenesis 5 (4): 511-4, 1984.
- Kraemer KH, Lee MM, Andrews AD, et al.: The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Dermatol 130 (8): 1018-21, 1994.
- Blankenburg S, König IR, Moessner R, et al.: Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: a case-control study. Carcinogenesis 26 (6): 1085-90, 2005.
- Xu Y, Jiao G, Wei L, et al.: Current evidences on the XPG Asp1104His polymorphism and melanoma susceptibility: a meta-analysis based on case-control studies. Mol Genet Genomics 290 (1): 273-9, 2015.
- Walpole S, Pritchard AL, Cebulla CM, et al.: Comprehensive Study of the Clinical Phenotype of Germline BAP1 Variant-Carrying Families Worldwide. J Natl Cancer Inst 110 (12): 1328-1341, 2018.
- Harbour JW, Onken MD, Roberson ED, et al.: Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 330 (6009): 1410-3, 2010.
- Wiesner T, Obenauf AC, Murali R, et al.: Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 43 (10): 1018-21, 2011.
- Soura E, Eliades PJ, Shannon K, et al.: Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counseling. J Am Acad Dermatol 74 (3): 411-20; quiz 421-2, 2016.
- Njauw CN, Kim I, Piris A, et al.: Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One 7 (4): e35295, 2012.
- Abdel-Rahman MH, Pilarski R, Cebulla CM, et al.: Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet 48 (12): 856-9, 2011.
- Carbone M, Ferris LK, Baumann F, et al.: BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med 10: 179, 2012.
- Testa JR, Cheung M, Pei J, et al.: Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 43 (10): 1022-5, 2011.
- Popova T, Hebert L, Jacquemin V, et al.: Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 92 (6): 974-80, 2013.
- Aoude LG, Wadt K, Bojesen A, et al.: A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One 8 (8): e72144, 2013.
- O'Shea SJ, Robles-Espinoza CD, McLellan L, et al.: A population-based analysis of germline BAP1 mutations in melanoma. Hum Mol Genet 26 (4): 717-728, 2017.
- Lalloo F, Kulkarni A, Chau C, et al.: Clinical practice guidelines for the diagnosis and surveillance of BAP1 tumour predisposition syndrome. Eur J Hum Genet 31 (11): 1261-1269, 2023.
- Wadt K, Choi J, Chung JY, et al.: A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 25 (6): 815-8, 2012.
- Rai K, Pilarski R, Cebulla CM, et al.: Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet 89 (3): 285-94, 2016.
- Cheung M, Kadariya Y, Talarchek J, et al.: Germline BAP1 mutation in a family with high incidence of multiple primary cancers and a potential gene-environment interaction. Cancer Lett 369 (2): 261-5, 2015.
- Walpole S, Hayward NK, Pritchard AL, et al.: Microsimulation Model for Evaluating the Cost-Effectiveness of Surveillance in BAP1 Pathogenic Variant Carriers. JCO Clin Cancer Inform 5: 143-154, 2021.
- Gillanders E, Juo SH, Holland EA, et al.: Localization of a novel melanoma susceptibility locus to 1p22. Am J Hum Genet 73 (2): 301-13, 2003.
- Walker GJ, Indsto JO, Sood R, et al.: Deletion mapping suggests that the 1p22 melanoma susceptibility gene is a tumor suppressor localized to a 9-Mb interval. Genes Chromosomes Cancer 41 (1): 56-64, 2004.
- Teerlink C, Farnham J, Allen-Brady K, et al.: A unique genome-wide association analysis in extended Utah high-risk pedigrees identifies a novel melanoma risk variant on chromosome arm 10q. Hum Genet 131 (1): 77-85, 2012.
- Höiom V, Tuominen R, Hansson J: Genome-wide linkage analysis of Swedish families to identify putative susceptibility loci for cutaneous malignant melanoma. Genes Chromosomes Cancer 50 (12): 1076-84, 2011.
- Tuominen R, Jönsson G, Enerbäck C, et al.: Investigation of a putative melanoma susceptibility locus at chromosome 3q29. Cancer Genet 207 (3): 70-4, 2014.
- Potrony M, Puig-Butille JA, Farnham JM, et al.: Genome-wide linkage analysis in Spanish melanoma-prone families identifies a new familial melanoma susceptibility locus at 11q. Eur J Hum Genet 26 (8): 1188-1193, 2018.
- Gudbjartsson DF, Sulem P, Stacey SN, et al.: ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat Genet 40 (7): 886-91, 2008.
- Maccioni L, Rachakonda PS, Scherer D, et al.: Variants at chromosome 20 (ASIP locus) and melanoma risk. Int J Cancer 132 (1): 42-54, 2013.
- Kanetsky PA, Swoyer J, Panossian S, et al.: A polymorphism in the agouti signaling protein gene is associated with human pigmentation. Am J Hum Genet 70 (3): 770-5, 2002.
- Landi MT, Kanetsky PA, Tsang S, et al.: MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J Natl Cancer Inst 97 (13): 998-1007, 2005.
- McMeniman EK, Duffy DL, Jagirdar K, et al.: The interplay of sun damage and genetic risk in Australian multiple and single primary melanoma cases and controls. Br J Dermatol 183 (2): 357-366, 2020.
- Bishop DT, Demenais F, Iles MM, et al.: Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet 41 (8): 920-5, 2009.
- Brown KM, Macgregor S, Montgomery GW, et al.: Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat Genet 40 (7): 838-40, 2008.
- Falchi M, Bataille V, Hayward NK, et al.: Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat Genet 41 (8): 915-9, 2009.
- Fernandez LP, Milne RL, Pita G, et al.: SLC45A2: a novel malignant melanoma-associated gene. Hum Mutat 29 (9): 1161-7, 2008.
- Kocarnik JM, Park SL, Han J, et al.: Replication of associations between GWAS SNPs and melanoma risk in the Population Architecture Using Genomics and Epidemiology (PAGE) Study. J Invest Dermatol 134 (7): 2049-52, 2014.
- Law MH, Bishop DT, Lee JE, et al.: Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat Genet 47 (9): 987-95, 2015.
- Gerstenblith MR, Shi J, Landi MT: Genome-wide association studies of pigmentation and skin cancer: a review and meta-analysis. Pigment Cell Melanoma Res 23 (5): 587-606, 2010.
- Athanasiadis EI, Antonopoulou K, Chatzinasiou F, et al.: A Web-based database of genetic association studies in cutaneous melanoma enhanced with network-driven data exploration tools. Database (Oxford) 2014: , 2014.
- Ohta M, Berd D, Shimizu M, et al.: Deletion mapping of chromosome region 9p21-p22 surrounding the CDKN2 locus in melanoma. Int J Cancer 65 (6): 762-7, 1996.
- Teerlink CC, Huff C, Stevens J, et al.: A Nonsynonymous Variant in the GOLM1 Gene in Cutaneous Malignant Melanoma. J Natl Cancer Inst 110 (12): 1380-1385, 2018.
- Box NF, Duffy DL, Chen W, et al.: MC1R genotype modifies risk of melanoma in families segregating CDKN2A mutations. Am J Hum Genet 69 (4): 765-73, 2001.
- Scherer D, Nagore E, Bermejo JL, et al.: Melanocortin receptor 1 variants and melanoma risk: a study of 2 European populations. Int J Cancer 125 (8): 1868-75, 2009.
- Kanetsky PA, Panossian S, Elder DE, et al.: Does MC1R genotype convey information about melanoma risk beyond risk phenotypes? Cancer 116 (10): 2416-28, 2010.
- Tagliabue E, Fargnoli MC, Gandini S, et al.: MC1R gene variants and non-melanoma skin cancer: a pooled-analysis from the M-SKIP project. Br J Cancer 113 (2): 354-63, 2015.
- Williams PF, Olsen CM, Hayward NK, et al.: Melanocortin 1 receptor and risk of cutaneous melanoma: a meta-analysis and estimates of population burden. Int J Cancer 129 (7): 1730-40, 2011.
- Puig-Butillé JA, Carrera C, Kumar R, et al.: Distribution of MC1R variants among melanoma subtypes: p.R163Q is associated with lentigo maligna melanoma in a Mediterranean population. Br J Dermatol 169 (4): 804-11, 2013.
- Pasquali E, García-Borrón JC, Fargnoli MC, et al.: MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer 136 (3): 618-31, 2015.
- Duffy DL, Box NF, Chen W, et al.: Interactive effects of MC1R and OCA2 on melanoma risk phenotypes. Hum Mol Genet 13 (4): 447-61, 2004.
- Davies JR, Randerson-Moor J, Kukalizch K, et al.: Inherited variants in the MC1R gene and survival from cutaneous melanoma: a BioGenoMEL study. Pigment Cell Melanoma Res 25 (3): 384-94, 2012.
- Peña-Vilabelda MM, García-Casado Z, Requena C, et al.: Clinical characteristics of patients with cutaneous melanoma according to variants in the melanocortin 1 receptor gene. Actas Dermosifiliogr 105 (2): 159-71, 2014.
- Taylor NJ, Reiner AS, Begg CB, et al.: Inherited variation at MC1R and ASIP and association with melanoma-specific survival. Int J Cancer 136 (11): 2659-67, 2015.
- Lira FE, Podlipnik S, Potrony M, et al.: Inherited MC1R variants in patients with melanoma are associated with better survival in women. Br J Dermatol 182 (1): 138-146, 2020.
- Dwyer T, Stankovich JM, Blizzard L, et al.: Does the addition of information on genotype improve prediction of the risk of melanoma and nonmelanoma skin cancer beyond that obtained from skin phenotype? Am J Epidemiol 159 (9): 826-33, 2004.
- Han J, Kraft P, Colditz GA, et al.: Melanocortin 1 receptor variants and skin cancer risk. Int J Cancer 119 (8): 1976-84, 2006.
- Cust AE, Goumas C, Vuong K, et al.: MC1R genotype as a predictor of early-onset melanoma, compared with self-reported and physician-measured traditional risk factors: an Australian case-control-family study. BMC Cancer 13: 406, 2013.
- Duffy DL, Lee KJ, Jagirdar K, et al.: High naevus count and MC1R red hair alleles contribute synergistically to increased melanoma risk. Br J Dermatol 181 (5): 1009-1016, 2019.
- Pellegrini C, Botta F, Massi D, et al.: MC1R variants in childhood and adolescent melanoma: a retrospective pooled analysis of a multicentre cohort. Lancet Child Adolesc Health 3 (5): 332-342, 2019.
- Wendt J, Mueller C, Rauscher S, et al.: Contributions by MC1R Variants to Melanoma Risk in Males and Females. JAMA Dermatol 154 (7): 789-795, 2018.
- van der Poel LAJ, Bergman W, Gruis NA, et al.: The role of MC1R gene variants and phenotypical features in predicting high nevus count. Melanoma Res 30 (5): 511-514, 2020.
- Demenais F, Mohamdi H, Chaudru V, et al.: Association of MC1R variants and host phenotypes with melanoma risk in CDKN2A mutation carriers: a GenoMEL study. J Natl Cancer Inst 102 (20): 1568-83, 2010.
- Fargnoli MC, Gandini S, Peris K, et al.: MC1R variants increase melanoma risk in families with CDKN2A mutations: a meta-analysis. Eur J Cancer 46 (8): 1413-20, 2010.
- Helsing P, Nymoen DA, Rootwelt H, et al.: MC1R, ASIP, TYR, and TYRP1 gene variants in a population-based series of multiple primary melanomas. Genes Chromosomes Cancer 51 (7): 654-61, 2012.
- Fargnoli MC, Fargnoli MC, Pike K, et al.: MC1R variants increase risk of melanomas harboring BRAF mutations. J Invest Dermatol 128 (10): 2485-90, 2008.
- Guida M, Strippoli S, Ferretta A, et al.: Detrimental effects of melanocortin-1 receptor (MC1R) variants on the clinical outcomes of BRAF V600 metastatic melanoma patients treated with BRAF inhibitors. Pigment Cell Melanoma Res 29 (6): 679-687, 2016.
- Hacker E, Olsen CM, Kvaskoff M, et al.: Histologic and Phenotypic Factors and MC1R Status Associated with BRAF(V600E), BRAF(V600K), and NRAS Mutations in a Community-Based Sample of 414 Cutaneous Melanomas. J Invest Dermatol 136 (4): 829-837, 2016.
- Thomas NE, Edmiston SN, Kanetsky PA, et al.: Associations of MC1R Genotype and Patient Phenotypes with BRAF and NRAS Mutations in Melanoma. J Invest Dermatol 137 (12): 2588-2598, 2017.
- Goldstein AM, Xiao Y, Sampson J, et al.: Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum Mol Genet 26 (24): 4886-4895, 2017.
- Yokoyama S, Woods SL, Boyle GM, et al.: A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480 (7375): 99-103, 2011.
- Sturm RA, Fox C, McClenahan P, et al.: Phenotypic characterization of nevus and tumor patterns in MITF E318K mutation carrier melanoma patients. J Invest Dermatol 134 (1): 141-9, 2014.
- Berwick M, MacArthur J, Orlow I, et al.: MITF E318K's effect on melanoma risk independent of, but modified by, other risk factors. Pigment Cell Melanoma Res 27 (3): 485-8, 2014.
- Ciccarese G, Dalmasso B, Bruno W, et al.: Clinical, pathological and dermoscopic phenotype of MITF p.E318K carrier cutaneous melanoma patients. J Transl Med 18 (1): 78, 2020.
- Guhan SM, Artomov M, McCormick S, et al.: Cancer risks associated with the germline MITF(E318K) variant. Sci Rep 10 (1): 17051, 2020.
- Carlo M, Schroeder A, Mukherjee Y, et al.: Germline mutations in hereditary renal cell carcinoma (RCC) genes among 32,240 patients with all cancer types. [Abstract] J Clin Oncol 41 (16): 4559, 2023. Also available online. Last accessed June 18, 2024.
- Cancer risks in BRCA2 mutation carriers. The Breast Cancer Linkage Consortium. J Natl Cancer Inst 91 (15): 1310-6, 1999.
- Moran A, O'Hara C, Khan S, et al.: Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer 11 (2): 235-42, 2012.
- van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al.: Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet 42 (9): 711-9, 2005.
- Kadouri L, Temper M, Grenader T, et al.: Absence of founder BRCA1 and BRCA2 mutations in cutaneous malignant melanoma patients of Ashkenazi origin. Fam Cancer 8 (1): 29-32, 2009.
- Gumaste PV, Penn LA, Cymerman RM, et al.: Skin cancer risk in BRCA1/2 mutation carriers. Br J Dermatol 172 (6): 1498-506, 2015.
- Johansson PA, Nathan V, Bourke LM, et al.: Evaluation of the contribution of germline variants in BRCA1 and BRCA2 to uveal and cutaneous melanoma. Melanoma Res 29 (5): 483-490, 2019.
- Bui AN, LeBoeuf NR, Nambudiri VE: Skin cancer risk in CHEK2 mutation carriers. J Eur Acad Dermatol Venereol 35 (2): 353-359, 2021.
- Weischer M, Heerfordt IM, Bojesen SE, et al.: CHEK2*1100delC and risk of malignant melanoma: Danish and German studies and meta-analysis. J Invest Dermatol 132 (2): 299-303, 2012.
- Hatton JN, Sargen MR, Frone MN, et al.: Spectrum and Incidence of Skin Cancer among Individuals with Li-Fraumeni Syndrome. J Invest Dermatol 142 (9): 2534-2537.e1, 2022.
- Nieuwenburg SA, Adan F, Ruijs MWG, et al.: Cumulative risk of skin cancer in patients with Li-Fraumeni syndrome. Fam Cancer 19 (4): 347-351, 2020.
- Lesueur F, Easton DF, Renault AL, et al.: First international workshop of the ATM and cancer risk group (4-5 December 2019). Fam Cancer 21 (2): 211-227, 2022.
- Taylor AM, Lam Z, Last JI, et al.: Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet 87 (3): 199-208, 2015.
- Hall MJ, Bernhisel R, Hughes E, et al.: Germline Pathogenic Variants in the Ataxia Telangiectasia Mutated (ATM) Gene are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev Res (Phila) 14 (4): 433-440, 2021.
- Mester J, Eng C: When overgrowth bumps into cancer: the PTEN-opathies. Am J Med Genet C Semin Med Genet 163C (2): 114-21, 2013.
- Pilarski R, Eng C: Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J Med Genet 41 (5): 323-6, 2004.
- Eng C: PTEN Hamartoma Tumor Syndrome (PHTS). In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed March 6, 2024.
- Pilarski R, Burt R, Kohlman W, et al.: Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst 105 (21): 1607-16, 2013.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 1.2023. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free registration. Last accessed June 24, 2024.
- Bubien V, Bonnet F, Brouste V, et al.: High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J Med Genet 50 (4): 255-63, 2013.
- Tan MH, Mester JL, Ngeow J, et al.: Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18 (2): 400-7, 2012.
- Nieuwenhuis MH, Kets CM, Murphy-Ryan M, et al.: Cancer risk and genotype-phenotype correlations in PTEN hamartoma tumor syndrome. Fam Cancer 13 (1): 57-63, 2014.
- Greene MH: The genetics of hereditary melanoma and nevi. 1998 update. Cancer 86 (11 Suppl): 2464-77, 1999.
- Olsen CM, Neale RE, Green AC, et al.: Independent validation of six melanoma risk prediction models. J Invest Dermatol 135 (5): 1377-84, 2015.
- Cho E, Rosner BA, Feskanich D, et al.: Risk factors and individual probabilities of melanoma for whites. J Clin Oncol 23 (12): 2669-75, 2005.
- Fears TR, Guerry D, Pfeiffer RM, et al.: Identifying individuals at high risk of melanoma: a practical predictor of absolute risk. J Clin Oncol 24 (22): 3590-6, 2006.
- Hampel H, Bennett RL, Buchanan A, et al.: A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: referral indications for cancer predisposition assessment. Genet Med 17 (1): 70-87, 2015.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Melanoma: Cutaneous. Version 3.2022. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2022. Available online with free registration. Last accessed June 24, 2024.
- Kefford RF, Newton Bishop JA, Bergman W, et al.: Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol 17 (10): 3245-51, 1999.
- Hansson J, Bergenmar M, Hofer PA, et al.: Monitoring of kindreds with hereditary predisposition for cutaneous melanoma and dysplastic nevus syndrome: results of a Swedish preventive program. J Clin Oncol 25 (19): 2819-24, 2007.
- Salerni G, Carrera C, Lovatto L, et al.: Benefits of total body photography and digital dermatoscopy ("two-step method of digital follow-up") in the early diagnosis of melanoma in patients at high risk for melanoma. J Am Acad Dermatol 67 (1): e17-27, 2012.
- Freedberg KA, Geller AC, Miller DR, et al.: Screening for malignant melanoma: A cost-effectiveness analysis. J Am Acad Dermatol 41 (5 Pt 1): 738-45, 1999.
- Pissa M, Lapins J, Sköldmark C, et al.: Melanoma-specific survival before and after inclusion in a familial melanoma dermatologic surveillance program in CDKN2A mutation carriers and non-carriers. J Eur Acad Dermatol Venereol 37 (2): 284-292, 2023.
- Tucker MA, Fraser MC, Goldstein AM, et al.: A natural history of melanomas and dysplastic nevi: an atlas of lesions in melanoma-prone families. Cancer 94 (12): 3192-209, 2002.
- National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version 3.2024. Plymouth Meeting, Pa: National Comprehensive Cancer Network, 2023. Available online with free registration. Last accessed April 11, 2024.
- Overbeek KA, Levink IJM, Koopmann BDM, et al.: Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 71 (6): 1152-1160, 2022.
- Goggins M, Overbeek KA, Brand R, et al.: Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 69 (1): 7-17, 2020.
- Vasen H, Ibrahim I, Ponce CG, et al.: Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J Clin Oncol 34 (17): 2010-9, 2016.
Síndromes relacionados con tipos raros de cáncer de piel
Síndrome de Brooke-Spiegler, tricoepitelioma múltiple familiar y cilindromatosis familiar
El síndrome de Brooke-Spiegler (SBS), la cilindromatosis familiar y el tricoepitelioma múltiple familiar (MFT) son síndromes autosómicos dominantes con características clínicas superpuestas y variación alélica.[1] Las características del SBS abarcan varios tumores apendiculares en la piel, como los cilindromas (tumores de las células madre del folículo piloso), los tricoepiteliomas (tumores del folículo piloso) y espiradenomas (tumores benignos de la glándula sudorípara). El TMF se caracteriza por tumores de piel no malignos, sobre todo tricoepiteliomas, y la citocromatosis familiar que se manifiesta de manera predominante como cilindromas cutáneos. El inicio de los tumores para estos síndromes suele ocurrir en la niñez tardía o la adolescencia temprana, lo que indica una influencia hormonal.[2] Hay indicios de mayor gravedad en mujeres que en hombres. La radiación UV parece ser un factor inicial importante para los cilindromas. En la cilindromatosis familiar, los sitios tumorales característicos son el cuero cabelludo (81 % de portadores), el tronco (69 % de portadores) y el área púbica (42 % de portadores).[3] Otros tumores que se pueden asociar con estos síndromes son los tumores de glándula parótida, los adenomas de células basales y los carcinomas de células basales. Para obtener más información sobre el SBS, consultar el Cuadro 3 Síndromes de carcinoma de células basales.
Se piensa que estos síndromes representan manifestaciones fenotípicas diferentes de la misma enfermedad, porque se han identificado variantes patogénicas de CYLD en16q12-q13 en personas con cada uno de estos síndromes.[4] La penetrancia de las variantes patogénicas de CYLD es del 60 % al 100 %.[3,5] En un estudio, el 85 % de las familias con SBS, el 100 % de las familias con cilindromatosis familiar y solo el 44 % de las familias con MFT, tenían variantes patogénicas de CYLD.[6] Otro locus para MFT se ubicó en 9p21, pero el gen para este locus no se conoce todavía.[7]
Dado el potencial de agrandamiento progresivo, el abordaje preferido para los cilindromas es la ablación cuando los tumores son pequeños y de fácil manejo. La electrocirugía o la cirugía micrográfica de Mohs son opciones de terapia, aunque la extirpación de lesiones grandes quizás exija injertos cutáneos para obtener cicatrización.[8] Los tricoepiteliomas y los espiradenomas por lo general no crecen mucho; de manera que no exigen intervención después de confirmar el diagnóstico por biopsia, a menos que causen pinzamiento de estructuras vitales. Cuando se indica tratamiento las opciones válidas son electrocirugía o terapia láser.[8] No se recomienda la radioterapia para el tratamiento de ninguno de estos tumores por el potencial de aumentar la inducción tumoral.
Nivel de evidencia: 4
Carcinoma sebáceo
Síndrome de Muir-Torre y síndrome de Lynch
Las neoplasias sebáceas cutáneas a veces se asocian con el síndrome de Muir-Torre (MTS). Se han descrito múltiples tipos de tumores sebáceos, como los adenomas sebáceos, los epiteliomas, los carcinomas, los queratoacantomas y los CCB con diferenciación sebácea, en el MTS. Este síndrome es una variación del síndrome de cáncer colorrectal no polipósico o síndrome de Lynch, que produce aparición sincrónica o metacrónica de por lo menos una neoplasia cutánea sebácea y una neoplasia visceral. Las neoplasias viscerales pueden originarse a nivel digestivo (colon y recto, estómago, intestino delgado, hígado y vías biliares) o urogenital (endometrio y vejiga). Estas neoplasias malignas suelen presentar un fenotipo menos maligno que los casos equivalentes de tumores no relacionados con el MTS.[9,10] El MTS se hereda de manera autosómica dominante y tiene penetrancia y expresividad variables. En un metanálisis se describieron las siguientes frecuencias de asociación entre el MTS con variantes patogénicas en genes de reparación de errores de emparejamiento: MLH1 (15 %), MSH2 (62 %), MSH6 (8 %) y PMS2 (1 %).[11,12,13,14,15,16,17] En un estudio de 36 lesiones sebáceas que incluyó carcinomas sebáceos, adenomas sebáceos y sebaceomas, el 38,9 % de las lesiones no tenía una o más proteínas de reparación de emparejamiento en el análisis inmunohistoquímico (IHC).[18] De las 10 personas que no contaban con la tinción para una o más proteínas de reparación de emparejamiento, 5 contaban con pruebas genéticas que confirmaban el diagnóstico de síndrome de Lynch. En 220 personas con diagnóstico confirmado de síndrome de Lynch (por prueba genética), el 95 % de las pruebas de inmunohistoquímica y el 90 % de las pruebas de inestabilidad microsatelital en 328 lesiones cutáneas fueron compatibles con la variante patogénica.[17] Para obtener más información en inglés, consultar la sección IHC en Genetics of Colorectal Cancer. Estos resultados indican que la detección sistemática mediante pruebas inmunohistoquímicas de las lesiones sebáceas quizás sea útil para identificar personas con síndrome de Lynch. En un estudio independiente, en 173 lesiones sebáceas cutáneas se analizaron las proteínas del síndrome de Lynch por inmunohistoquímica en pacientes que desconocían si tenían un síndrome de Lynch.[19] Se encontraron resultados anormales en las pruebas de inmunohistoquímica en el 53 % (92/173) de las lesiones sebáceas cutáneas. De 101 pacientes, 47 fueron derivados a pruebas genéticas y las completaron. Entre ellos, 15 % tenía una variante patogénica de un gen relacionado con el síndrome de Lynch (más frecuentemente el gen MSH2). Para obtener más información en inglés, consultar la sección Lynch Syndrome en Genetics of Colorectal Cancer.
Si bien se ha indicado con frecuencia que la hiperplasia sebácea no se asocia con el MTS, cualquier lesión sebácea con características histológicas atípicas o dificultad para la clasificación justifica exploración inmediata de los antecedentes del paciente y su historia familiar. Se justifica atención especial cuando los pacientes con neoplasias sebáceas se derivan al genetista médico o gastroenterólogo para las evaluaciones del síndrome de Lynch. Si bien la mayoría de los pacientes con MTS reciben el diagnóstico de una neoplasia maligna visceral antes de presentar una neoplasia maligna cutánea, el 22 % de los pacientes primero presenta la neoplasia sebácea cutánea. Esta situación brinda la oportunidad de detectar las neoplasias viscerales.[20] El diagnóstico de MTS se basa en criterios clínicos, pero se apoya en la tinción inmunohistoquímica para MSH2, MLH1 y MSH6, lo que puede servir como método de detección sistemática antes de las pruebas genéticomoleculares.[12,14,15,16,21] Se recomienda que los pacientes y familiares en quienes se sospeche MTS reciban asesoramiento y pruebas genéticas y sigan los programas de detección sistemática para neoplasias malignas viscerales.
Nivel de evidencia: 3
Síndrome de Lynch y otros factores de riesgo de cáncer de piel
En múltiples estudios se ha indicado que las personas con síndrome de Lynch tienen un riesgo más alto de neoplasias cutáneas. En un estudio de 607 personas con síndrome de Lynch (mediana de edad en el momento de la prueba genética de 50 años) se encontró que el 21 % tenía una neoplasia cutánea, casi un tercio de aquellos con más de un tipo histológico.[22] El tipo de neoplasia cutánea más comúnmente diagnosticada en estos pacientes fue el CCB (9 % de los pacientes). Las siguientes neoplasias en orden de frecuencia fueron los adenomas sebáceos y los carcinomas de células escamosas (CCE) en el 7,2 % y 5,8 % de los pacientes, respectivamente. Otras neoplasias de piel en estos pacientes fueron melanoma, carcinoma sebáceo, epiteliomas sebáceos o sebaceomas, queratoacantoma y carcinoma de células de Merkel. Estos hallazgos cutáneos fueron más frecuentes en hombres (oportunidad relativa [OR], 2,44), peronas de mayor edad, personas blancas y portadores de variantes patogénicas de MLH1 o MSH2/EPCAM. Una debilidad de este estudio fue que los investigadores no evaluaron en las lesiones cutáneas la presencia de alteración en la reparación de emparejamiento porque los cánceres de piel son comunes en la población general. En un estudio donde se analizó la incidencia de cáncer de piel en pacientes con síndrome de Lynch se indica un incremento en los carcinomas sebáceos y CCB.[23] Se necesitan otros estudios para determinar si las variantes patogénicas en los genes del síndrome de Lynch causan CCE.[17]
Leiomiomatosis hereditaria y carcinoma de células renales
Aunque los tumores de músculo liso cutáneos (leiomiomas) no son en sí mismos una forma de cáncer de piel, la presencia de múltiples leiomiomas cutáneos se relaciona con cáncer de células renales (CCR) en pacientes que tienen un síndrome hereditario conocido como leiomiomatosis hereditaria y cáncer de células renales (HLRCC). Este síndrome se vincula con variantes patogénicas germinales del gen FH. Los leiomiomas cutáneos se presentan en forma de pápulas y nódulos firmes, rosados o de color marrón rojizo, en el tronco y las extremidades y, en ocasiones, en la cara. Aparecen a una mediana de edad de 25 años (intervalo de edad, 10–47 años) y tienden a aumentar de tamaño y número con la edad. Las lesiones son sensibles al tacto suave o al frío y, con menos frecuencia, son dolorosas. La intensidad del dolor se correlaciona con la gravedad del compromiso cutáneo.[24] La presencia de varios leiomiomas cutáneos se relaciona con la HLRCC hasta que se demuestre lo contrario y debe dar lugar a un estudio genético; un leiomioma solitario requiere un análisis cuidadoso de la historia familiar. La HLRCC también se asocia con fibroides uterinos de importancia clínica y cánceres renales de gran malignidad. Para obtener más información, consultar Leiomiomatosis hereditaria y cáncer de células renales.
Referencias:
- Bowen S, Gill M, Lee DA, et al.: Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol 124 (5): 919-20, 2005.
- Burrows NP, Jones RR, Smith NP: The clinicopathological features of familial cylindromas and trichoepitheliomas (Brooke-Spiegler syndrome): a report of two families. Clin Exp Dermatol 17 (5): 332-6, 1992.
- Rajan N, Langtry JA, Ashworth A, et al.: Tumor mapping in 2 large multigenerational families with CYLD mutations: implications for disease management and tumor induction. Arch Dermatol 145 (11): 1277-84, 2009.
- Young AL, Kellermayer R, Szigeti R, et al.: CYLD mutations underlie Brooke-Spiegler, familial cylindromatosis, and multiple familial trichoepithelioma syndromes. Clin Genet 70 (3): 246-9, 2006.
- Welch JP, Wells RS, Kerr CB: Ancell-Spiegler cylindromas (turban tumours) and Brooke-Fordyce Trichoepitheliomas: evidence for a single genetic entity. J Med Genet 5 (1): 29-35, 1968.
- Saggar S, Chernoff KA, Lodha S, et al.: CYLD mutations in familial skin appendage tumours. J Med Genet 45 (5): 298-302, 2008.
- Harada H, Hashimoto K, Ko MS: The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol 107 (1): 41-3, 1996.
- Rajan N, Trainer AH, Burn J, et al.: Familial cylindromatosis and brooke-spiegler syndrome: a review of current therapeutic approaches and the surgical challenges posed by two affected families. Dermatol Surg 35 (5): 845-52, 2009.
- Schwartz RA, Torre DP: The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol 33 (1): 90-104, 1995.
- Cohen PR, Kohn SR, Kurzrock R: Association of sebaceous gland tumors and internal malignancy: the Muir-Torre syndrome. Am J Med 90 (5): 606-13, 1991.
- Cerosaletti KM, Lange E, Stringham HM, et al.: Fine localization of the Nijmegen breakage syndrome gene to 8q21: evidence for a common founder haplotype. Am J Hum Genet 63 (1): 125-34, 1998.
- Mangold E, Pagenstecher C, Leister M, et al.: A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet 41 (7): 567-72, 2004.
- Mathiak M, Rütten A, Mangold E, et al.: Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol 26 (3): 338-43, 2002.
- Mangold E, Rahner N, Friedrichs N, et al.: MSH6 mutation in Muir-Torre syndrome: could this be a rare finding? Br J Dermatol 156 (1): 158-62, 2007.
- Arnold A, Payne S, Fisher S, et al.: An individual with Muir-Torre syndrome found to have a pathogenic MSH6 gene mutation. Fam Cancer 6 (3): 317-21, 2007.
- Murphy HR, Armstrong R, Cairns D, et al.: Muir-Torre Syndrome: expanding the genotype and phenotype--a further family with a MSH6 mutation. Fam Cancer 7 (3): 255-7, 2008.
- Aziz S, O'Sullivan H, Heelan K, et al.: Characterization of sebaceous and non-sebaceous cutaneous manifestations in patients with lynch syndrome: a systematic review. Fam Cancer 22 (2): 167-175, 2023.
- Plocharczyk EF, Frankel WL, Hampel H, et al.: Mismatch repair protein deficiency is common in sebaceous neoplasms and suggests the importance of screening for Lynch syndrome. Am J Dermatopathol 35 (2): 191-5, 2013.
- Kattapuram M, Shabet C, Austin S, et al.: A retrospective cohort study of genetic referral and diagnosis of Lynch syndrome in patients with cutaneous sebaceous lesions. Fam Cancer 22 (3): 295-301, 2023.
- Akhtar S, Oza KK, Khan SA, et al.: Muir-Torre syndrome: case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol 41 (5 Pt 1): 681-6, 1999.
- Entius MM, Keller JJ, Drillenburg P, et al.: Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res 6 (5): 1784-9, 2000.
- Zhong CS, Horiguchi M, Uno H, et al.: Clinical factors associated with skin neoplasms in individuals with Lynch syndrome in a longitudinal observational cohort. J Am Acad Dermatol 88 (6): 1282-1290, 2023.
- Adan F, Crijns MB, Zandstra WSE, et al.: Cumulative risk of skin tumours in patients with Lynch syndrome. Br J Dermatol 179 (2): 522-523, 2018.
- Toro JR, Nickerson ML, Wei MH, et al.: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 73 (1): 95-106, 2003.
Aspectos psicosociales del melanoma familiar
Introducción
En esta sección se revisan las publicaciones que tratan sobre la reducción del riesgo y las conductas de detección temprana en personas con riesgo elevado de melanoma por una historia familiar de esta enfermedad, y en personas de familias que se hicieron pruebas de variantes patogénicas de riesgo alto de melanoma. En la revisión también se aborda la percepción del riesgo y la comunicación en personas con riesgo elevado de melanoma.
Interés y aceptación de las pruebas genéticas de riesgo de melanoma
En pocos estudios se examinó la motivación y el interés en la prueba genética para el riesgo de melanoma. En resumen, los resultados son los siguientes:
- Interés alto, pero no universal, en la prueba genética.[1,2,3]
- Beneficios articulados de la prueba en personas con riesgo elevado.[1,2,4]
- Cierta carencia de un análisis de las posibles limitaciones de las pruebas o de las razones para abstenerse de la prueba.[1,2,3]
En Australia, en un estudio cualitativo (N = 40) se encontró que casi todos los participantes con historia familiar significativa con melanoma tenían interés en someterse a la prueba genética.[2,5] Los participantes favorecieron el uso de pruebas genéticas como medio de obtener información sobre la susceptibilidad al melanoma en sus hijos, aumentar la comprensión de su propio riesgo, promover la investigación sobre el melanoma y aumentar la motivación para adoptar un comportamiento de protección solar.
En un estudio holandés se examinó el interés en la prueba para CDKN2A (variante patogénica Leiden p16). De las 510 cartas enviadas a miembros de 18 familias con resultados positivos para Leiden p16 de un consultorio para lesiones pigmentadas en Leiden University Medical Center de los Países Bajos, 488 personas respondieron asistiendo a una consulta para someterse a un examen físico y otros 15 familiares acompañaron a estas personas. De estas personas que consultaron, 403 fueron elegibles para asesoramiento genético. En total, 184 personas asistieron al asesoramiento y 141 optaron por hacerse la prueba genética. Después de la consulta de orientación, 94 personas devolvieron un cuestionario completo. La edad avanzada predijo un interés más alto en la prueba genética; las razones para hacerse las pruebas fueron conocer el riesgo personal (57 %) y conocer el riesgo de un hijo o hija que porta la variante patogénica (69 %). La mayoría de los participantes (88 %) opinaron que la prueba genética contribuiría al diagnóstico dentro de su familia. Sin embargo, algunas personas (40 %) comentaron que no esperaban recibir información sobre riesgo de cáncer de páncreas, y la mitad de los participantes (49 %) expresaron aumento de la preocupación sobre la posibilidad de presentar cáncer de páncreas.[3] Por último, en Arizona se hizo un estudio cualitativo en 22 personas con una historia familiar significativa de melanoma, y nadie optó por someterse a una prueba genética aunque se les ofreció dicha oportunidad.[4]
En un estudio australiano de 121 personas con una historia familiar significativa de melanoma, los participantes completaron cuestionarios antes del asesoramiento y la prueba genética.[5] El grado de angustia (específica por melanoma o sufrimiento en general) fueron muy bajos en esta población. Los factores predictivos más importantes de la angustia fueron los antecedentes personales de melanoma, las preocupaciones sobre las repercusiones del melanoma en la familia, una disposición por buscar información (estilo vigilante), una importancia percibida de que la exposición al sol causa melanoma y no tener descendencia.
Pruebas en la niñez
En una revisión sistemática se analizaron 3 estudios que examinaban las actitudes de los participantes sobre si se debía realizar una prueba genética de melanoma hereditario en menores.[6] La mayoría de los participantes estaban a favor de hacer la prueba de riesgo de melanoma en menores. Muchos participantes que apoyaban las pruebas en menores creían que la prueba genética podría aumentar el grado de sensibilización sobre el riesgo de melanoma y promover las conductas de protección solar.
Conocimiento y reducción del riesgo en personas con un riesgo familiar elevado de melanoma
Se han llevado a cabo varios estudios en los que se examina la reducción del riesgo mediante la adopción de conductas de protección solar (incluso el uso de protector solar y ropa protectora, y la búsqueda de sombra) en personas con historia familiar de melanoma. En general, estos estudios indican una adopción y mantenimiento irregulares de estos comportamientos. La mayoría de estos estudios se han llevado a cabo con poblaciones de consultorios que podrían ser más propensas a implementar conductas de reducción del riesgo y de detección que otras personas con un perfil de riesgo similar de la población general.[7]
En una población sueca se entrevistaron 87 adultos jóvenes con nevos displásicos con respecto a sus conductas de protección solar. El 70 % estimó que su riesgo de melanoma era igual o inferior al riesgo general de la población sueca, y un tercio notificó que tomaba el sol a menudo.[8] En otro estudio, se evaluaron a 229 familiares de primer grado (FPG) de pacientes con melanoma que asistieron a consulta clínica; aquellos familiares de mayor edad, las mujeres, y aquellos con una mayor confianza en su habilidad para protegerse del sol tenían una probabilidad más alta de protegerse, pero incluso así, su comportamiento era irregular.[9] En otro estudio en los Estados Unidos se examinaron las conductas de protección solar en 100 FPG de pacientes con melanoma de un consultorio y se encontró que menos de un tercio de los pacientes usaban, de rutina, protector solar y el grado de cuidado se relacionó con un nivel de escolaridad más alto, mayor confianza en su habilidad para protegerse del sol y una percepción de mayor riesgo de melanoma. La gravedad percibida del melanoma y la eficacia de la respuesta no se relacionaron con la adopción de conductas de protección solar.[10]
En un estudio centrado en 68 menores (17 años o menos) sobrevivientes de melanoma, se demostró que, aunque las tasas generales de conductas de protección solar eran altas (cerca del 80 %), las tasas de quemaduras solares también fueron altas (49 %).[11] Los autores concluyeron que se necesitan varios tipos de conductas de protección solar en estas personas. Sin embargo, durante la adolescencia, hubo reducciones significativas en las conductas de protección solar, lo que indica una necesidad aún mayor de intervención en este grupo.
En otro estudio del Reino Unido se examinaron las tasas de quemaduras solares en 170 personas con historia familiar de melanoma y se compararon con las tasas de 140 controles emparejados por edad, sexo y ubicación geográfica. De los pacientes con historia familiar de melanoma, el 31 % notificó quemaduras solares en el verano anterior (en comparación con 41 % de los controles); las familias con melanoma notificaron mejores conductas de protección solar que los controles en general. En los controles y en los pacientes con historia familiar de melanoma, los varones más jóvenes tenían más probabilidades de notificar quemaduras solares recientes; además, en los controles y los pacientes con una historia familiar de melanoma, los familiares con síndrome de lunar atípico y confianza en su capacidad para prevenir el melanoma mostraron un mejor grado de protección solar.[12]
En un estudio retrospectivo internacional de una cohorte grande (547 familias, 2047 personas) de melanoma familiar (GenoMEL) se examinó la exposición solar de manera longitudinal y las conductas de protección solar en diferentes cohortes de nacimiento. Los investigadores encontraron que la exposición solar había sido similar en todas las cohortes de nacimiento. El uso de protector solar aumentó a lo largo de la vida de los participantes y el uso fue más frecuente en las cohortes de nacimiento más recientes. No se encontraron diferencias en los antecedentes de exposición al sol, uso de protector solar o de cámaras de bronceado entre las personas con melanoma y aquellas que no tenían este tipo de cáncer. Sin embargo, las conductas de protección solar aumentaron después de que los participantes recibieron el diagnóstico de melanoma.[13]
En un estudio cualitativo de 20 FPG de pacientes con melanoma de un consultorio de riesgo alto vinculado a la University of Arizona, se identificaron necesidades insatisfechas en cuanto a la comunicación sobre el estado de riesgo durante la consulta médica, entre ellas, una mayor congruencia en la comunicación, educación para los pacientes sobre la importancia de la historia familiar para definir el estado de riesgo, y necesidades y deseo de recibir un asesoramiento más profundo (por ejemplo, la aplicación repetida de protector solar y el uso de ropa con factor de protección ultravioleta).[14]
En un estudio prospectivo se examinó el interés en los desenlaces conductuales y psicosociales a los 3 meses relacionados con la divulgación de los resultados de la prueba de variantes patogénicas de riesgo alto para el melanoma en 19 personas (3 portadores de CDKN2A).[15] Todos los portadores de variantes y solo 4 de quienes no portaban variantes, tenían una historia familiar de melanoma. El estado de portador no afectó la percepción del riesgo, el sufrimiento ni las conductas de protección solar.
Comportamientos de detección en personas con un riesgo familiar elevado de melanoma
En varios estudios se examinaron las conductas de detección temprana en personas con un riesgo más alto de melanoma. En una muestra estadounidense de 404 hermanos y hermanas de una población de pacientes con melanoma atendida en un consultorio, solo el 42 % de las personas había asistido a una consulta de dermatología en su vida; el 62 % se había hecho un autoexamen cutáneo; el 27 % se había sometido a un examen médico de la piel; y solo el 54 % usaban protector solar de manera rutinaria. El sexo femenino se relacionó con mayor uso de protector solar y las personas mayores de 50 años tenían más probabilidades de haberse sometido a un examen médico de la piel. Se encontró una relación fuerte entre asistir a consulta de dermatología y los 3 desenlaces (autoexamen de la piel, examen médico y uso de protector solar).[16] En un estudio de los Estados Unidos con 229 FPG remitidos por pacientes atendidos en un consultorio, cerca de la mitad (55 %) notificó haberse realizado un examen médico total de la piel, y algunos más (71 %) notificaron haberse realizado un autoexamen de la piel. Los factores de predicción comunes para el examen de la piel (médico o autoadministrado) fueron la recomendación médica y una percepción de pocas barreras para la detección.[9] Cabe destacar, que el 14 % de las personas de la muestra no le había comunicado al médico de atención primaria sobre el diagnóstico de melanoma en su hermano o hermana. En un estudio de los Estados Unidos, se observó que la mitad (53 %) de los FPG nunca se habían sometido a un examen médico de toda la piel para detección y solo el 27 % había recibido una recomendación médica de hacerse un examen de detección. El cumplimiento con la detección temprana se relacionó con los siguientes aspectos: mayor nivel de educación, más factores de riesgo de melanoma, recomendación de proveedor de atención de la salud sobre someterse a un examen de detección, riesgo percibido de melanoma y gravedad percibida de melanoma. Fue menos probable que los progenitores de pacientes con melanoma se sometieran a detección en comparación con hermanos o hijos de los pacientes.[17] En un estudio de los Estados Unidos se examinaron las intenciones de someterse a un examen médico de la piel y de realizarse un autoexamen cutáneo en FPG de personas con diagnóstico de melanoma que no se habían sometido antes a vigilancia cutánea. Los factores de predicción de las intenciones fueron la percepción de beneficios y barreras para los exámenes de detección y el apoyo familiar para los exámenes de detección, pero no el conocimiento de la frecuencia recomendada de estos exámenes.[18]
En un estudio transversal de Australia con 120 personas de familias con una variante patogénica del gen CDKN2A conocida, se encontró que en los últimos 12 meses, el 50 % notificó haberse hecho por lo menos 4 autoexámenes de la piel, y 43 % se había sometido al menos a 1 examen clínico de la piel. En contraste, el 15 % no se había realizado un autoexamen de la piel en los últimos 12 meses, y el 27 % nunca se había hecho un examen clínico de la piel. Las conductas sobre exámenes de detección del cáncer de piel se correlacionaron con tener antecedentes de melanoma, una recomendación médica e intenciones conductuales más fuertes. Otras correlaciones para el autoexamen cutáneo fueron la confianza en la habilidad para protegerse del sol, la percepción de la eficacia del tratamiento con melanoma y la angustia específica por el melanoma. El riesgo percibido de presentar melanoma no se relacionó de manera significativa con las conductas de detección del cáncer de piel.[19]
En una revisión sistemática se proporcionó un resumen descriptivo de los resultados de comportamiento (conductas de protección solar, autoexamen de piel y examen clínico de la piel) relacionados con la prueba genética del riesgo de melanoma hereditario. Aunque se presentaron algunas diferencias entre los estudios, en más de la mitad de los estudios se encontró que una variante patogénica hereditaria se asociaba con aumento del cumplimiento (o las intenciones) con las recomendaciones de prevención y detección del melanoma. No se encontró disminución en las conductas de protección solar en las personas no portadoras de la variante.[20]
Estudios de intervención
Algunos estudios de intervención se enfocan en el conocimiento del melanoma, la protección solar y los exámenes de detección en familiares de pacientes con melanoma. En un estudio de una población atendida en un consultorio, un grupo de hermanos y hermanas de pacientes se asignaron al azar a una intervención que incluía mensajes telefónicos y materiales impresos personalizados sobre las recomendaciones de reducción del riesgo y exámenes de detección. El grupo de atención habitual recibió una recomendación estándar durante la consulta médica sobre la notificación del diagnóstico a los familiares del paciente. En el grupo de intervención se observaron mejoras en el conocimiento sobre el melanoma, confianza en que debían solicitar una consulta por dermatología para someterse a un examen de detección, y mejora más significativa en las prácticas de autoexamen de la piel en comparación con los participantes de control después de 12 meses; en ambos grupos se duplicó el número de exámenes médicos después de 12 meses; no hubo cambios en el uso de protector solar en ninguno de los grupos.[21]
En otro estudio, se asignó al azar a 443 familiares de pacientes con melanoma a un grupo de atención genérica y uno de una intervención adaptada que constaba ya fuera de 3 correos impresos (no personalizados y personalizados) o 1 sesión telefónica de orientación (no personalizada y personalizada). En general, en el grupo de intervención personalizada se observó un aumento casi al doble de la frecuencia de los exámenes clínicos de toda la piel en una consulta de atención primaria en comparación con la intervención genérica. Sin embargo, no se observaron diferencias en los autoexámenes cutáneos en los grupos de intervención. En contraste con el estudio previo, en el que no se observaron mejoras en las conductas de protección solar,[21] fue significativamente más probable que los participantes en este estudio que recibieron la intervención personalizada notificaran mejoras de las conductas de protección solar en comparación con quienes no las recibieron.[22]
Resultados psicosociales del asesoramiento genético y la prueba genética
En una revisión sistemática se examinaron varios resultados psicosociales en personas de alto riesgo sometidas a una prueba genética de melanoma y que participaron en estudios cuyos resultados se publicaron entre 1995 y 2020.[6] Los resultados psicosociales incluyeron ansiedad generalizada, depresión, angustia específica por melanoma, preocupación por melanoma y cáncer pancreático, y arrepentimiento sobre la prueba genética. En general, los grados de angustia (general y específica por melanoma) fueron bajos y subclínicos. La ansiedad disminuyó con el tiempo, tanto en portadores como en no portadores. Los resultados de las preocupaciones específicas por cáncer fueron contradictorios en los estudios al compararlos entre portadores y no portadores, personas afectadas y no afectadas y cambió de la preocupación con el tiempo. En los estudios se observó que las personas no portadoras sobrestimaron el riesgo percibido de cáncer en comparación con la estimación objetiva. En general, se observaron grados bajos de arrepentimiento de la prueba genética. Se describió una tendencia a enumerar más beneficios que limitaciones de la prueba genética.
Referencias:
- Bränström R, Kasparian NA, Affleck P, et al.: Perceptions of genetic research and testing among members of families with an increased risk of malignant melanoma. Eur J Cancer 48 (16): 3052-62, 2012.
- Kasparian NA, Meiser B, Butow PN, et al.: Anticipated uptake of genetic testing for familial melanoma in an Australian sample: An exploratory study. Psychooncology 16 (1): 69-78, 2007.
- de Snoo FA, Riedijk SR, van Mil AM, et al.: Genetic testing in familial melanoma: uptake and implications. Psychooncology 17 (8): 790-6, 2008.
- Loescher LJ, Crist JD, Siaki LA: Perceived intrafamily melanoma risk communication. Cancer Nurs 32 (3): 203-10, 2009 May-Jun.
- Kasparian NA, Butow PN, Meiser B, et al.: High- and average-risk individuals' beliefs about, and perceptions of, malignant melanoma: an Australian perspective. Psychooncology 17 (3): 270-9, 2008.
- Primiero CA, Yanes T, Finnane A, et al.: A Systematic Review on the Impact of Genetic Testing for Familial Melanoma II: Psychosocial Outcomes and Attitudes. Dermatology 237 (5): 816-826, 2021.
- Shuk E, Burkhalter JE, Baguer CF, et al.: Factors associated with inconsistent sun protection in first-degree relatives of melanoma survivors. Qual Health Res 22 (7): 934-45, 2012.
- Bergenmar M, Brandberg Y: Sunbathing and sun-protection behaviors and attitudes of young Swedish adults with hereditary risk for malignant melanoma. Cancer Nurs 24 (5): 341-50, 2001.
- Manne S, Fasanella N, Connors J, et al.: Sun protection and skin surveillance practices among relatives of patients with malignant melanoma: prevalence and predictors. Prev Med 39 (1): 36-47, 2004.
- Azzarello LM, Dessureault S, Jacobsen PB: Sun-protective behavior among individuals with a family history of melanoma. Cancer Epidemiol Biomarkers Prev 15 (1): 142-5, 2006.
- Glenn BA, Bastani R, Chang LC, et al.: Sun protection practices among children with a family history of melanoma: a pilot study. J Cancer Educ 27 (4): 731-7, 2012.
- Newton Bishop JA, Gruis NA: Genetics: what advice for patients who present with a family history of melanoma? Semin Oncol 34 (6): 452-9, 2007.
- Lacson JCA, Zamani SA, Froes LAR, et al.: Birth cohort-specific trends of sun-related behaviors among individuals from an international consortium of melanoma-prone families. BMC Public Health 21 (1): 692, 2021.
- Loescher LJ, Crist JD, Cranmer L, et al.: Melanoma high-risk families' perceived health care provider risk communication. J Cancer Educ 24 (4): 301-7, 2009.
- Christensen KD, Roberts JS, Shalowitz DI, et al.: Disclosing individual CDKN2A research results to melanoma survivors: interest, impact, and demands on researchers. Cancer Epidemiol Biomarkers Prev 20 (3): 522-9, 2011.
- Geller AC, Emmons K, Brooks DR, et al.: Skin cancer prevention and detection practices among siblings of patients with melanoma. J Am Acad Dermatol 49 (4): 631-8, 2003.
- Azzarello LM, Jacobsen PB: Factors influencing participation in cutaneous screening among individuals with a family history of melanoma. J Am Acad Dermatol 56 (3): 398-406, 2007.
- Coups EJ, Manne SL, Jacobsen PB, et al.: Skin surveillance intentions among family members of patients with melanoma. BMC Public Health 11: 866, 2011.
- Kasparian NA, McLoone JK, Meiser B, et al.: Skin cancer screening behaviours among individuals with a strong family history of malignant melanoma. Br J Cancer 103 (10): 1502-9, 2010.
- Primiero CA, Yanes T, Finnane A, et al.: A Systematic Review on the Impact of Genetic Testing for Familial Melanoma I: Primary and Secondary Preventative Behaviours. Dermatology 237 (5): 806-815, 2021.
- Geller AC, Emmons KM, Brooks DR, et al.: A randomized trial to improve early detection and prevention practices among siblings of melanoma patients. Cancer 107 (4): 806-14, 2006.
- Manne S, Jacobsen PB, Ming ME, et al.: Tailored versus generic interventions for skin cancer risk reduction for family members of melanoma patients. Health Psychol 29 (6): 583-93, 2010.
Actualizaciones más recientes a este resumen (07 / 25 / 2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Melanoma
Se añadió texto para indicar que en un estudio en el que se realizó la secuenciación de POT1 en casi 3000 personas con melanoma y 3300 controles, se identificaron 43 variantes de secuencia, de inmediato se predijo que 6 de ellas eran patogénicas. En estudios funcionales, se encontró otras 9 variantes de POT1 con un efecto en la función proteínica. En general, el 0,5 % de las personas con melanoma eran portadoras de una variante potencialmente patogénica en el gen POT1 (se citó a Simonin-Wilmer et al. como referencia 126).
Se añadió texto para indicar que en un metanálisis de 9 estudios publicados se demostró que la variante MITF E318K es un alelo de riesgo moderado de melanoma (se citó a Guhan et al. como referencia 202). También se indicó que aunque en estudios anteriores se indicó una relación entre las variantes patogénicas de MITF y el aumento de riesgo del carcinoma de células renales, en estudios más grandes no se ha confirmado esta relación (se citó a Carlo et al. como referencia 203).
Se añadió texto para indicar que en estudios longitudinales y retrospectivos de personas con cáncer de piel y síndrome de Li-Fraumeni (SLF) se encontró un riesgo acumulado del 36,3 % al 44,6 % para cualquier tipo de cáncer de piel a la edad de 70 años (se citó a Nieuwenburg et al. como referencia 213). También se añadió texto sobre los riesgos de melanoma, carcinoma de células basales y carcinoma de células escamosas en las personas con SLF.
El Consejo editorial del PDQ sobre la genética del cáncer es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre la genética del cáncer de piel. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre la genética del cáncer, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
- Si el artículo se debe analizar en una reunión del consejo.
- Si conviene añadir texto acerca del artículo.
- Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Genética del cáncer de piel son:
- Kathleen A. Calzone, PhD, RN, AGN-BC, FAAN (National Cancer Institute)
- Joanne Marie Jeter, MD (City of Hope)
- Kelly Carter Nelson, MD, FAAD (M.D. Anderson Cancer Center at University of Texas)
- Suzanne C. O'Neill, PhD (Georgetown University)
- Susan K. Peterson, PhD, MPH (University of Texas, M.D. Anderson Cancer Center)
- John M. Quillin, PhD, MPH, MS (Virginia Commonwealth University)
- Charite Ricker, MS, CGC (University of Southern California)
- Amanda Ewart Toland, PhD, FACMG (The Ohio State University)
- Catharine Wang, PhD, MSc (Boston University School of Public Health)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre la genética del cáncer emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
PDQ® sobre la genética del cáncer. PDQ Genética del cáncer de piel. Bethesda, MD: National Cancer Institute. Actualización: <MM/DD/YYYY>. Disponible en: https://www.cancer.gov/espanol/tipos/piel/pro/genetica-cancer-de-piel-pdq. Fecha de acceso: <MM/DD/YYYY>.
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
La información en estos resúmenes no se debe utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Comuníquese con el Instituto Nacional del Cáncer
Para obtener más información sobre las opciones para comunicarse con el NCI, incluso la dirección de correo electrónico, el número telefónico o el chat, consultar la página del Servicio de Información de Cáncer del Instituto Nacional del Cáncer.
Última revisión: 2024-07-25

