Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil (PDQ®) : Tratamiento - información para profesionales de salud [NCI]
Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre el tumor teratoide rabdoide atípico del sistema nervioso central infantil
Los tumores primarios de encéfalo, también llamados tumores encefálicos o cerebrales, como los tumores teratoides rabdoides atípicos, son un grupo diverso de enfermedades que juntas constituyen los tumores sólidos más comunes en la niñez. Los resúmenes de PDQ sobre el tratamiento de los tumores de encéfalo infantiles se organizan, sobre todo, de acuerdo con la clasificación de tumores del sistema nervioso establecida por la Organización Mundial de la Salud.[1,2] Los tumores de encéfalo se clasifican según sus características histológicas, pero los hallazgos de análisis inmunohistoquímicos, citogenéticos y genético-moleculares, así como las mediciones de la actividad mitótica se utilizan cada vez más para diagnosticar y clasificar estos tumores. La ubicación del tumor, su grado de diseminación y la edad en el momento del diagnóstico son factores importantes que afectan el tratamiento y el pronóstico.[3,4,5] Para obtener una descripción de la clasificación de los tumores del sistema nervioso y un enlace al resumen de tratamiento correspondiente a cada tipo de tumor encefálico, consultar Resumen de la clasificación de los tumores de encéfalo y médula espinal infantiles.
El tumor teratoide rabdoide atípico (TTRA) del sistema nervioso central (SNC) es un tumor raro, muy maligno desde el punto de vista clínico, que la mayoría de las veces afecta a niños de 3 años o menos, pero que también se presenta en niños de más edad y en adultos. Alrededor de la mitad de los TTRA se forman en la fosa posterior.[6] La evaluación diagnóstica incluye imágenes por resonancia magnética (IRM) del eje encefalomedular y un examen del líquido cefalorraquídeo lumbar. El TTRA se ha vinculado con variantes somáticas y germinales de SMARCB1 y, con menor frecuencia, en SMARCA4; ambos actúan como genes supresores de tumores.[7] No se dispone de un tratamiento estándar basado en la evidencia para los niños con TTRA. Está en evaluación un tratamiento multimodal en ensayos clínicos controlados que incluye cirugía, quimioterapia (incluso quimioterapia de dosis alta) y radioterapia.
Según la comprensión biológica actual, el TTRA forma parte de un grupo más amplio de tumores rabdoides. En este resumen, la expresión TTRA se refiere solo a los tumores del SNC, y el término tumor rabdoide contempla la posibilidad de que los tumores se formen en el SNC o fuera de este. A menos que se indique lo contrario en el texto, este resumen se refiere a los TTRA del SNC.
Los niños y adolescentes sobrevivientes de cáncer necesitan un seguimiento minucioso, ya que es posible que los efectos secundarios del tratamiento del cáncer persistan o se presenten meses o años después de este. Para obtener información específica sobre la incidencia, el tipo y la vigilancia de los efectos tardíos en niños y adolescentes sobrevivientes de cáncer, consultar Efectos tardíos del tratamiento anticanceroso en la niñez.
Incidencia
Es difícil determinar la incidencia exacta de los TTRA del SNC, ya que este tumor es raro y recién se reconoció a partir de 1996.[8]
En dos estudios prospectivos realizados en América del Norte por el Children's Cancer Group y el Pediatric Oncology Group, la revisión retrospectiva reveló que casi el 10 % de los niños de 3 años o menos en el momento del diagnóstico con tumores de encéfalo tenía TTRA.[9]
En un estudio realizado en Taiwán, se encontró que los TTRA representaban el 26 % de los tumores primitivos o embrionarios en niños menores de 3 años.[10]
En el Austrian Brain Tumor Registry (período de inscripción de pacientes, 1996–2006), se confirmó que los TTRA eran el sexto tumor cerebral más común en 311 niños recién diagnosticados (6,1 %); con una incidencia máxima durante los 2 primeros años de vida.[11]
Se desconoce la incidencia en pacientes de más edad. Sin embargo, en el Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry (AT/RT Registry), 12 de 42 pacientes (29 %) tenían más de 36 meses en el momento del diagnóstico.[12]
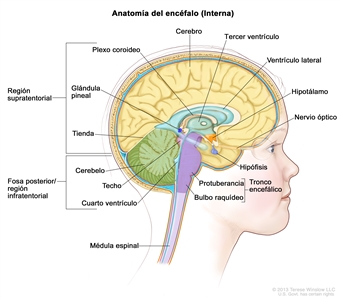
Características anatómicas
Cuadro clínico inicial
El TTRA infantil es un tumor maligno desde el punto de vista clínico que se presenta sobre todo en niños menores de 3 años, pero que también se observa en niños de más edad y en adultos.[13,14]
Cerca de la mitad de los TTRA surgen en la fosa posterior, aunque es posible que se presenten en cualquier parte del SNC.[6,9] Los tumores de la fosa posterior en ocasiones se forman en el ángulo pontocerebelar o más cerca de la línea media. También se ha observado compromiso de nervios craneales individuales.[15]
Dado que los TTRA crecen rápido, los pacientes a menudo tienen una evolución de síntomas progresivos bastante breve, medida en días o semanas. Los signos y síntomas dependen de la ubicación del tumor. Los pacientes jóvenes con tumores de la fosa posterior suelen presentar síntomas relacionados con hidrocefalia, como los siguientes:
Cefaleas en la madrugada.
Vómitos.
Letargo.
Aumento de la circunferencia de la cabeza.
En ocasiones, también presentan ataxia, regresión de las habilidades motoras o síntomas localizados relacionados con la disfunción de los nervios craneales.
Los datos de un registro indican que del 25 % al 30 % de los pacientes tienen diseminación de la enfermedad.[5,12,16] La diseminación se produce, por lo general, a través de vías leptomeníngeas y deja semillas de metástasis en la columna vertebral y en otras áreas del encéfalo. Hasta un 35 % de los pacientes exhiben variantes de la línea germinal y quizás sean propensos a tener tumores sincrónicos multifocales.[17,18,19,20]
Evaluación diagnóstica
Todos los pacientes en quienes se sospecha un TTRA se deben someter a IRM del encéfalo y la columna vertebral. A menos que esté contraindicado desde el punto de vista médico, se debe realizar una inspección del líquido cefalorraquídeo para evaluar si hay indicios de tumor. En ocasiones, también se somete a los pacientes a una ecografía renal para detectar tumores sincrónicos. Asimismo, se indican pruebas de la línea germinal.
No se puede distinguir de manera confiable los TTRA de otros tumores de encéfalo malignos a partir de la historia clínica o la evaluación radiográfica solas. Es necesaria la cirugía para obtener tejido y confirmar el diagnóstico. La tinción inmunohistoquímica para determinar la pérdida de expresión de la proteína SMARCB1 también se usa para confirmar el diagnóstico.[21,22] El análisis de metilación se ha convertido en un complemento importante para confirmar el subtipo TTRA.[3,4]
Pronóstico
Los factores pronósticos que afectan la supervivencia de pacientes con TTRA no están plenamente delineados.
Los siguientes son los factores conocidos que están relacionados con un desenlace adverso:
La mayor parte de los datos publicados sobre los desenlaces de los pacientes con TTRA se obtuvieron en series pequeñas y son de carácter retrospectivo. En los estudios retrospectivos iniciales se notificó una supervivencia promedio desde el diagnóstico de solo 12 meses, aproximadamente.[8,9,13,25,27] En un informe retrospectivo, la supervivencia general (SG) a 2 años fue mejor en los pacientes que se sometieron a una resección macroscópica total que en aquellos con una resección subtotal. Sin embargo, el efecto de la radioterapia en la supervivencia fue menos claro en este estudio.[25]
Hay informes que dan cuenta de sobrevivientes a largo plazo.[28] En particular, se notificó una mejora de la supervivencia en aquellos que recibieron terapia multimodal intensiva.[16,19]
Los niños de 3 años y más con TTRA que recibieron irradiación craneoespinal después de la cirugía y quimioterapia de dosis altas con un alquilante tuvieron una mejor supervivencia, en comparación con los pacientes menores de 3 años. En este informe, la incidencia de metástasis leptomeníngeas también fue más alta en los lactantes.[29]
En un estudio prospectivo de 25 niños con TTRA que recibieron terapia multimodal intensiva, incluso radioterapia y quimioterapia intratecal, la tasa notificada de supervivencia sin progresión a 2 años fue del 53 % y la tasa de SG fue del 70 %.[30]
En los pacientes del ensayo prospectivo ACNS0333 (NCT00653068), la tasa de supervivencia sin complicaciones a 4 años fue del 37 % y la tasa de SG a 4 años fue del 43 %.[31]
En la serie prospectiva del European Rhabdoid Registry, se observó que los pacientes de 1 año o más con una designación de subgrupo TTRA-tirosinasa (TYR) presentaron una tasa de SG a 5 años del 71 %, mientras que aquellos menores de 1 año con una designación diferente a la del subgrupo TTRA TYR tuvieron una tasa de supervivencia muy precaria.[5] Estos datos se confirmaron en otros dos ensayos.[26]
Referencias:
Louis DN, Perry A, Wesseling P, et al.: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23 (8): 1231-1251, 2021.
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
Federico A, Thomas C, Miskiewicz K, et al.: ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol 143 (6): 697-711, 2022.
Lu VM, Di L, Eichberg DG, et al.: Age of diagnosis clinically differentiates atypical teratoid/rhabdoid tumors diagnosed below age of 3 years: a database study. Childs Nerv Syst 37 (4): 1077-1085, 2021.
Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
Dho YS, Kim SK, Cheon JE, et al.: Investigation of the location of atypical teratoid/rhabdoid tumor. Childs Nerv Syst 31 (8): 1305-11, 2015.
Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.
Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.
Ho DM, Hsu CY, Wong TT, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99 (5): 482-8, 2000.
Woehrer A, Slavc I, Waldhoer T, et al.: Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116 (24): 5725-32, 2010.
Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.
Burger PC, Yu IT, Tihan T, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22 (9): 1083-92, 1998.
Lutterbach J, Liegibel J, Koch D, et al.: Atypical teratoid/rhabdoid tumors in adult patients: case report and review of the literature. J Neurooncol 52 (1): 49-56, 2001.
Lobón-Iglesias MJ, Andrianteranagna M, Han ZY, et al.: Imaging and multi-omics datasets converge to define different neural progenitor origins for ATRT-SHH subgroups. Nat Commun 14 (1): 6669, 2023.
Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.
Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.
Bourdeaut F, Lequin D, Brugières L, et al.: Frequent hSNF5/INI1 germline mutations in patients with rhabdoid tumor. Clin Cancer Res 17 (1): 31-8, 2011.
Seeringer A, Reinhard H, Hasselblatt M, et al.: Synchronous congenital malignant rhabdoid tumor of the orbit and atypical teratoid/rhabdoid tumor--feasibility and efficacy of multimodal therapy in a long-term survivor. Cancer Genet 207 (9): 429-33, 2014.
Nemes K, Clément N, Kachanov D, et al.: The extraordinary challenge of treating patients with congenital rhabdoid tumors-a collaborative European effort. Pediatr Blood Cancer 65 (6): e26999, 2018.
Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.
Margol AS, Judkins AR: Pathology and diagnosis of SMARCB1-deficient tumors. Cancer Genet 207 (9): 358-64, 2014.
Kordes U, Gesk S, Frühwald MC, et al.: Clinical and molecular features in patients with atypical teratoid rhabdoid tumor or malignant rhabdoid tumor. Genes Chromosomes Cancer 49 (2): 176-81, 2010.
Dufour C, Beaugrand A, Le Deley MC, et al.: Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118 (15): 3812-21, 2012.
Lafay-Cousin L, Hawkins C, Carret AS, et al.: Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48 (3): 353-9, 2012.
Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin Cancer Res 27 (10): 2879-2889, 2021.
Athale UH, Duckworth J, Odame I, et al.: Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31 (9): 651-63, 2009.
Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.
Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.
Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.
Reddy AT, Strother DR, Judkins AR, et al.: Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children's Oncology Group Trial ACNS0333. J Clin Oncol 38 (11): 1175-1185, 2020.
Características biológicas del tumor teratoide rabdoide atípico del sistema nervioso central infantil
El tumor teratoide rabdoide atípico (TTRA) del sistema nervioso central (SNC) infantil se describió por primera vez como una entidad clínica aislada en 1987 [1] a partir de sus características patológicas y genéticas distintivas. Antes de eso, se clasificaba con mayor frecuencia como meduloblastoma, tumor neuroectodérmico primitivo del SNC (TNEP del SNC) o carcinoma del plexo coroideo. La Organización Mundial de la Salud (OMS) clasifica el TTRA como una neoplasia embrionaria de grado IV.[2]
Desde el punto de vista histológico, el TTRA es morfológicamente heterogéneo, suele contener capas de células epiteliales grandes con abundante citoplasma eosinofílico y células rabdoides dispersas, por lo general con componentes acompañantes de células neuroectodérmicas primitivas (células pequeñas, redondas y azuladas), células mesenquimatosas o células gliales.[3]
La tinción inmunohistoquímica de marcadores epiteliales (citoqueratina o antígeno de la membrana epitelial), proteína ácida fibrilar glial, sinaptofisina (o neurofilamento) y músculo liso (desmina) en ocasiones ayuda a identificar la heterogeneidad de diferenciación, pero variará según la composición celular.[4] Si bien no están presentes en todos los TTRA, las células rabdoides expresarán vimentina, antígeno de la membrana epitelial y actina de músculo liso.
La prueba inmunohistoquímica para la proteína SMARCB1 es útil para establecer el diagnóstico de TTRA. Se observa una pérdida de tinción SMARCB1 en las células neoplásicas, pero permanece en las células que no son neoplásicas (por ejemplo, las células endoteliales vasculares).[5,6,7]
El TTRA es un tumor de crecimiento rápido que tiene un índice de rotulación MIB-1 del 50 % al 100 %.[8]
Características genómicas del tumor teratoide rabdoide atípico del sistema nervioso central
GenesSMARCB1ySMARCA4
El tumor teratoide rabdoide atípico (TTRA) fue el primer tumor encefálico primario infantil en el que se identificó el gen SMARCB1 y se lo propuso como gen oncosupresor.[9] El gen SMARCB1 tiene alteraciones genómicas en la mayoría de los tumores rabdoides, incluso en las neoplasias malignas rabdoides del SNC, renales y extrarrenales.[9] El SMARCB1 es un componente del complejo de reestructuración de la cromatina de SWItch y sacarosa no fermentable (SWI/SNF).[10]
Los casos raros de tumores rabdoides que expresan SMARCB1 y carecen de variantes de SMARCB1 también se relacionaron con variantes somáticas o germinales de SMARCA4, otro miembro del complejo de reestructuración de la cromatina SWI/SNF.[7,11,12]
Con menor frecuencia, se describieron tumores negativos para SMARCA4 (pero que retienen SMARCB1).[7,11,12] La pérdida de tinción de SMARCB1 o SMARCA4 es un marcador que define el TTRA.
En la clasificación de 2021 de la OMS se define el TTRA por la presencia de alteraciones en SMARCB1 o SMARCA4. Los tumores con características histológicas de TTRA que no tienen estas alteraciones genómicas se denominan tumores embrionarios del SNC con características rabdoides.[13]
A pesar de la ausencia de alteraciones genómicas recurrentes diferentes a las de SMARCB1 y SMARCA4,[14,15,16] se han identificado subconjuntos de TTRA relativamente específicos desde el punto de vista biológico.[17,18,19] En un estudio se identificaron tres subconjuntos específicos de TTRA mediante el empleo de matrices de metilación del DNA para 150 TTRA y matrices de expresión génica para 67 TTRA:[18]
TTRA-tirosinasa (TYR). Este subconjunto representa casi un tercio de los casos y se caracterizó por una expresión alta de marcadores melanosómicos como TYR (gen que codifica la tirosinasa). Los casos en este subconjunto fueron sobre todo infratentoriales; la mayoría de los casos se presentaron en niños de 0 a 1 año y exhibían pérdida del cromosoma 22q.[18] En los pacientes que tenían TTRA TYR, la media de supervivencia general (SG) fue de 37 meses en un grupo heterogéneo desde el punto de vista clínico (intervalo de confianza [IC] 95 %, 18–56 meses).[20] En una serie prospectiva del European Rhabdoid Registry (EU-RHAB), se observó que los pacientes de 1 año o más con TTRA TYR presentaban una tasa de SG a 5 años del 71 %, mientras que aquellos menores de 1 año con TTRA TYR tuvieron una tasa de supervivencia muy precaria.[21]
TTRA-erizo sónico (SHH). Este subconjunto representó cerca del 40 % de los casos y se caracterizó por una expresión alta de los genes de la vía del erizo sónico (sonic hedgehog [SHH]) (por ejemplo, GLI2 y MYCN). Los casos de este subconjunto se presentaron con una frecuencia similar a nivel supratentorial e infratentorial. Si bien la mayoría de los casos se manifestaron antes de los 2 años de edad, alrededor de un tercio de los casos se presentó entre los 2 y los 5 años.[18] En los pacientes con TTRA SHH, la media de SG fue de 16 meses (IC 95 %, 8–25 meses).[20]
En un estudio posterior, el subgrupo TTRA SHH se dividió a su vez en tres subtipos: SHH-1A, SHH-1B y SHH-2.[22] Los niños mayores de 3 años que albergaban la firma SHH-1B experimentaron los resultados más favorables.
TTRA MYC. Este subconjunto representa casi un cuarto de los casos y se caracterizó por una expresión alta de MYC. Los casos de TTRA MYC tienden a presentarse en el compartimento supratentorial. Si bien la mayoría de los casos de TTRA MYC se presentaron a los 5 años de edad, el TTRA MYC representó el subconjunto más común diagnosticado a la edad de 6 años o más. Las deleciones focales en SMARCB1 fueron el mecanismo más común de pérdida de SMARCB1 en este subconjunto.[18] En los pacientes con TTRA MYC, la media de SG fue de 13 meses (IC 95 %, 5–22 meses).[20]
La pérdida de la expresión de las proteínas SMARCB1 o SMARCA4 tiene importancia terapéutica, porque esta pérdida crea una dependencia de la actividad de EZH2 por parte de las células cancerosas.[23] En estudios preclínicos se observó que algunas líneas de xenoinjerto de TTRA con pérdida de SMARCB1 responden a los inhibidores de EZH2 presentando inhibición del crecimiento tumoral y, en ocasiones, regresión tumoral.[24,25] En un estudio del inhibidor de EZH2, tazemetostat, se observaron respuestas objetivas en pacientes adultos cuyos tumores exhibían pérdida de SMARCB1 o SMARCA4 (tumores rabdoides malignos fuera del SNC y sarcoma epitelioide).[26] Para obtener más información, consultar la sección Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil recidivante.
Tumor neuroepitelial cribiforme
El tumor neuroepitelial cribiforme presenta características genómicas y epigenómicas muy similares a las del TTRA TYR.[20] La clasificación de la OMS de 2021 incluye el tumor neuroepitelial cribiforme como entidad provisional. Al igual que el TTRA, el tumor neuroepitelial cribiforme se presenta en niños pequeños (mediana de edad, 1–2 años) y las células tumorales carecen de expresión de SMARCB1. Desde el punto histológico, el tumor neuroepitelial cribiforme se caracteriza por la presencia de cordones y cintas cribiformes, pero hay ausencia de células tumorales rabdoides con abundante citoplasma eosinófilo. Al igual que el TTRA TYR, se observa con frecuencia la expresión de tirosinasa. El pronóstico de los pacientes con tumor neuroepitelial cribiforme es más favorable que el de aquellos con TTRA TYR. En un estudio, solo se notificó una muerte entre 10 niños con tumor neuroepitelial cribiforme.[20]
Síndrome de predisposición a tumores rabdoides
Se definió de forma clara el síndrome de predisposición a tumores rabdoides (RTPS) que, por lo general, se relaciona con alteraciones germinales de SMARCB1 (y en menor medida con alteraciones germinales de SMARCA4).[9,27] El RTPS causado por alteraciones germinales de SMARCB1 se denomina RTPS tipo 1, mientras que el RTPS debido a una variante germinal de SMARCA4 se denomina RTPS tipo 2. La presentación sincrónica de tumor rabdoide maligno extracraneal (de riñón o tejido blando) y TTRA, tumores rabdoides malignos bilaterales renales, o tumores rabdoides malignos en dos o más hermanos, es muy indicativa de RTPS.
Este síndrome se manifiesta por una marcada predisposición a la presentación de tumores rabdoides malignos durante la lactancia y la niñez temprana. Se piensa que hasta un tercio de los TTRA surgen en el entorno de un RTPS; la mayoría de ellos se presentan en el primer año de vida. La neoplasia maligna más frecuente del RTPS que no afecta el SNC es el tumor rabdoide maligno de riñón, que también se observa durante la lactancia.[28,29]
En un estudio de 65 niños con tumores rabdoides se descubrió que 23 (35 %) de ellos tenían variantes de la línea germinal o deleciones de SMARCB1.[5] Los niños con alteraciones germinales de SMARCB1 presentaron el tumor a una edad más temprana que los casos esporádicos (mediana de edad, aproximadamente 5 meses vs. 18 meses) y era más probable que presentaran tumores sincrónicos y multifocales.[5] Se descubrió que uno de los progenitores era portador de la anomalía germinal de SMARCB1 en 7 de los 22 casos evaluados que mostraban alteraciones de la línea germinal, y que 4 de los progenitores portadores no estaban afectados por cánceres relacionados con SMARCB1.[5] Este hallazgo indica que la TTRA muestra un patrón de herencia autosómico dominante con penetrancia incompleta.
También se ha observado mosaicismo gonadal, como se ha demostrado en familias en las que varios hermanos están afectados por TTRA y presentan alteraciones idénticas de SMARCB1, pero en las que ambos progenitores carecen de una variante o deleción de SMARCB1.[5,6] Se indica el examen de detección de variantes germinales de SMARCB1 en niños diagnosticados con TTRA para asesorar a las familias sobre las implicaciones genéticas del diagnóstico de TTRA de sus hijos.[5] Se han publicado recomendaciones preliminares para la evaluación genética y el posterior examen presintomático de portadores de variantes no afectados (incluidos progenitores y hermanos de ambos sexos de pacientes afectados), que probablemente evolucionarán a medida que mejore el conocimiento del RTPS.[28,29,30] En pacientes con predisposición al TTRA, la resonancia magnética de cuerpo entero puede ayudar a identificar tumores rabdoides sincrónicos fuera del SNC.
Para obtener más información en inglés sobre el RTPS1 y SMARCB1, consultar Rhabdoid Tumor Predisposition Syndrome Type 1.
Referencias:
Rorke LB, Packer RJ, Biegel JA: Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg 85 (1): 56-65, 1996.
Oztek MA, Noda SM, Romberg EK, et al.: Changes to pediatric brain tumors in 2021 World Health Organization classification of tumors of the central nervous system. Pediatr Radiol 53 (3): 523-543, 2023.
Louis DN, Ohgaki H, Wiestler OD: WHO Classification of Tumours of the Central Nervous System. 4th rev.ed. IARC Press, 2016.
McLendon RE, Adekunle A, Rajaram V, et al.: Embryonal central nervous system neoplasms arising in infants and young children: a pediatric brain tumor consortium study. Arch Pathol Lab Med 135 (8): 984-93, 2011.
Eaton KW, Tooke LS, Wainwright LM, et al.: Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer 56 (1): 7-15, 2011.
Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56 (7): 1026-31, 2011.
Hasselblatt M, Gesk S, Oyen F, et al.: Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35 (6): 933-5, 2011.
Kleihues P, Louis DN, Scheithauer BW, et al.: The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61 (3): 215-25; discussion 226-9, 2002.
Biegel JA, Tan L, Zhang F, et al.: Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8 (11): 3461-7, 2002.
Biegel JA, Kalpana G, Knudsen ES, et al.: The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res 62 (1): 323-8, 2002.
Schneppenheim R, Frühwald MC, Gesk S, et al.: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86 (2): 279-84, 2010.
Hasselblatt M, Nagel I, Oyen F, et al.: SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol 128 (3): 453-6, 2014.
WHO Classification of Tumours Editorial Board, ed.: WHO Classification of Tumours: Central Nervous System Tumours. Vol. 6. 5th ed. IARC Press; 2021.
Lee RS, Stewart C, Carter SL, et al.: A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122 (8): 2983-8, 2012.
Kieran MW, Roberts CW, Chi SN, et al.: Absence of oncogenic canonical pathway mutations in aggressive pediatric rhabdoid tumors. Pediatr Blood Cancer 59 (7): 1155-7, 2012.
Hasselblatt M, Isken S, Linge A, et al.: High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosomes Cancer 52 (2): 185-90, 2013.
Torchia J, Picard D, Lafay-Cousin L, et al.: Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16 (5): 569-82, 2015.
Johann PD, Erkek S, Zapatka M, et al.: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29 (3): 379-93, 2016.
Upadhyaya SA, Robinson GW, Onar-Thomas A, et al.: Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional Trials. Clin Cancer Res 27 (10): 2879-2889, 2021.
Johann PD, Hovestadt V, Thomas C, et al.: Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 27 (4): 411-418, 2017.
Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
Federico A, Thomas C, Miskiewicz K, et al.: ATRT-SHH comprises three molecular subgroups with characteristic clinical and histopathological features and prognostic significance. Acta Neuropathol 143 (6): 697-711, 2022.
Wilson BG, Wang X, Shen X, et al.: Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18 (4): 316-28, 2010.
Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.
Kurmasheva RT, Sammons M, Favours E, et al.: Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer 64 (3): , 2017.
Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.
Biegel JA, Fogelgren B, Wainwright LM, et al.: Germline INI1 mutation in a patient with a central nervous system atypical teratoid tumor and renal rhabdoid tumor. Genes Chromosomes Cancer 28 (1): 31-7, 2000.
Frühwald MC, Nemes K, Boztug H, et al.: Current recommendations for clinical surveillance and genetic testing in rhabdoid tumor predisposition: a report from the SIOPE Host Genome Working Group. Fam Cancer 20 (4): 305-316, 2021.
Nemes K, Bens S, Bourdeaut F, et al.: Rhabdoid Tumor Predisposition Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., eds.: GeneReviews. University of Washington, Seattle, 1993-2024, pp. Available online. Last accessed December 15, 2023.
Foulkes WD, Kamihara J, Evans DGR, et al.: Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res 23 (12): e62-e67, 2017.
Información sobre los estadios del tumor teratoide rabdoide atípico del sistema nervioso central infantil
No hay ningún sistema de estadificación basado en la evidencia para el tumor teratoide rabdoide atípico del sistema nervioso central infantil. A efectos de tratamiento, los pacientes se clasifican con enfermedad recién diagnosticada o recidivante con diseminación al eje encefalomedular o sin esta.
Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil
Aún no se ha definido un tratamiento estándar basado en la evidencia para los niños con tumor teratoide rabdoide atípico (TTRA) del sistema nervioso central (SNC) recién diagnosticado. Dada la naturaleza sumamente maligna del tumor, la mayoría de los pacientes se tratan con terapia multimodal intensiva. Sin embargo, la edad temprana de la mayoría de los pacientes impone limitaciones a la intensidad del tratamiento, en particular, de la radioterapia.
Las siguientes son las opciones de tratamiento de los TTRA del SNC recién diagnosticados:
Cirugía, quimioterapia y radioterapia (terapia multimodal).
Cirugía, quimioterapia y radioterapia (terapia multimodal)
Es posible que el alcance de la resección quirúrgica afecte la supervivencia. Los datos del Central Nervous System Atypical Teratoid/Rhabdoid Tumor Registry (AT/RT Registry) indican que los pacientes sometidos a resección completa quizás tengan una mediana de supervivencia más alta. No obstante, la resección quirúrgica completa suele ser difícil debido a la naturaleza invasiva del tumor.[1]
La quimioterapia ha sido la principal terapia adyuvante para los niños muy pequeños con TTRA. En estudios de un grupo cooperativo, en los que se incluyó a niños menores de 36 meses, se demostró que la supervivencia fue precaria solo con regímenes quimioterapéuticos estándar.[2] El Children's Cancer Group notificó una tasa de supervivencia sin complicaciones (SSC) a 2 años del 14 % en 28 niños menores de 36 meses tratados con quimioterapia multifarmacológica.[3]
Los regímenes intensivos en los que se usan varias combinaciones quimioterapéuticas de dosis altas,[4][Nivel de evidencia C1]; [5,6][Nivel de evidencia C2] quimioterapia intratecal y radioterapia condujeron a una supervivencia prolongada en algunos pacientes.
Solo se han completado dos ensayos prospectivos de niños con TTRA del SNC. En un ensayo prospectivo institucional, los niños recibieron tratamiento con un protocolo modificado del Intergroup Rhabdomyosarcoma Study-III (IRS-III) mediante quimioterapia intratecal y radioterapia. Del subconjunto de 20 niños que completaron el tratamiento, la tasa de supervivencia sin progresión (SSP) a los 2 años fue del 53 %, y la tasa de supervivencia general (SG) fue del 70 %. La supervivencia fue mejor en los pacientes a los que se realizó una resección completa.[7][Nivel de evidencia C1] En el estudio del Children's Oncology Group (COG) ACNS0333 (NCT00653068) los pacientes recibieron tratamiento con quimioterapia de inducción intensiva, seguida de quimioterapia de dosis alta con rescate autógeno de células madre y radioterapia. La tasa de SSP a 4 años fue del 37 % y la de SG del 43 %.[8][Nivel de evidencia B4]
Se trató a 13 pacientes del AT/RT Registry con quimioterapia de dosis altas y rescate de células madre hematopoyéticas como parte del tratamiento inicial.[1] Según el último informe, 4 de estos pacientes (2 de los cuales también recibieron radiación), estaban vivos y sin progresión de la enfermedad entre 21,5 y 90 meses después del diagnóstico. De 15 niños evaluables (todos menores de 32 meses en el momento del diagnóstico) que participaron en un protocolo quimioterapéutico de Head Start III, 2 sobrevivieron por más de 47 meses.[9][Nivel de evidencia C1]
La radioterapia parece tener un efecto positivo en la supervivencia de los pacientes con TTRA.[10]
Evidencia (radioterapia):
De los 42 pacientes del AT/RT Registry, 13 (31 %) recibieron radioterapia además de quimioterapia como parte de su tratamiento primario.[1] El campo de radiación se dirigió al lecho del tumor primario en 9 niños, y al lecho del tumor y el eje craneoespinal en 4 niños.
La mediana de supervivencia de estos pacientes fue de 48 meses, en comparación con 16,75 meses de todos los pacientes del registro.
En una serie retrospectiva de 31 pacientes con TTRA del St. Jude Children's Research Hospital, se notificaron los siguientes desenlaces:[11]
La tasa de SSC a 2 años de los pacientes mayores de 3 años fue del 78 %; bastante mejor que la tasa de SSC del 11 % de los pacientes menores de 3 años.
Todos los pacientes sobrevivientes, salvo 1, (7 de 8) del grupo de mayor edad, recibieron irradiación craneoespinal y quimioterapia intensiva con trasplante de células madre hematopoyéticas.
Solo 3 de los 22 pacientes más jóvenes recibieron alguna forma de radioterapia y 2 de ellos permanecieron sin enfermedad.
En una revisión del registro Surveillance, Epidemiology, and End Results (SEER) Program, la radioterapia se relacionó con mejor supervivencia en niños menores de 3 años.[12]
En el European Registry de series de tumores rabdoides, se observaron los siguientes desenlaces:[13][Nivel de evidencia C1]
La radioterapia también se vinculó con una mejora de la supervivencia; la tasa de SG a 6 años fue del 66 % (± 0,1 %) en los pacientes que recibieron este tratamiento.
En una extensión de estas series se corroboró el beneficio significativo de la radioterapia.[14]
Evidencia (terapia multimodal):
En el estudio IRS-III se usó radioterapia, metotrexato intratecal, citarabina, hidrocortisona y quimioterapia sistémica multifarmacológica. Los resultados de esta serie retrospectiva pequeña fueron alentadores,[15,16] lo que condujo al primer estudio prospectivo de tratamiento multimodal para este grupo de pacientes.
A partir de la serie piloto previa, se realizó un ensayo multinstitucional prospectivo con niños recién diagnosticados con TTRA del SNC. El tratamiento se dividió en 5 fases: preirradiación, quimiorradiación, consolidación, mantenimiento y terapia de continuación. Se administró quimioterapia intratecal, alternando la ruta intralumbar y la intraventricular. La radioterapia fue focal (54 Gy) o craneoespinal (36 Gy, más refuerzo primario), según la edad del niño y el grado de la enfermedad en el momento del diagnóstico.[7]
La tasa de SSP a 2 años fue del 53 % (± 13 %) y la tasa de SG a 2 años fue del 70 % (± 10 %).
Los resultados fueron más favorables en niños mayores sometidos a resección macroscópica total y sin enfermedad metastásica en el momento de la presentación.
De los 8 niños sin enfermedad progresiva en el momento del informe, 6 habían recibido radioterapia conformada y 2 habían recibido radioterapia craneoespinal; 7 niños se sometieron a resección macroscópica total y solo 1 niño presentó enfermedad metastásica (este niño presentaba enfermedad persistente y estable 1,5 años después del diagnóstico).
El COG realizó un estudio prospectivo de un solo grupo de 65 niños. De esos niños, 54 eran menores de 36 meses y recibieron 2 cursos de metotrexato, ciclofosfamida, cisplatino y etopósido seguido de 3 cursos de dosis altas de carboplatino y tiotepa con apoyo de rescate de células madre periféricas. En los pacientes con enfermedad no diseminada, se ordenó la radioterapia focal al campo comprometido después de la inducción o la consolidación, dependiendo de la edad del paciente. En los pacientes con enfermedad diseminada, se recomendó, pero no se ordenó, la radioterapia craneoespinal al final del tratamiento.[8]
En todos los pacientes, la tasa de SSC a 4 años fue del 37 % y la tasa de SG a 4 años fue del 43 %.
De los niños menores de 36 meses en el momento del diagnóstico, la tasa de SSC a 4 años fue del 35 %, en comparación con el 6,4 % de una cohorte histórica de pacientes que recibieron quimioterapia sola (P < 0,0005).
En los 11 niños con edad de 36 meses o más en el momento del diagnóstico, la tasa de SSC a 4 años fue del 48 % y la tasa de SG a 4 años fue del 57 %.
La toxicidad de este régimen fue importante. Se notificaron 4 muertes relacionadas con el tratamiento (6 % de los pacientes), debido a sepsis, insuficiencia respiratoria o necrosis del SNC.
A partir de los 2 estudios que se resumieron antes, la terapia multimodal con cirugía, radioterapia y quimioterapia, parece ser el mejor tratamiento para optimizar la supervivencia de los niños con TTRA. Sin embargo, los efectos tóxicos pueden ser importantes, y todavía se debe determinar el régimen más eficaz y la secuencia óptima de las terapias.
Opciones de tratamiento en evaluación clínica
Tal vez haya ensayos clínicos terapéuticos de fase inicial para ciertos pacientes. Es posible que estos ensayos estén disponibles a través del Children's Oncology Group, el Pediatric Brain Tumor Consortium u otras entidades. La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de internet ClinicalTrials.gov.
Referencias:
Hilden JM, Meerbaum S, Burger P, et al.: Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22 (14): 2877-84, 2004.
Packer RJ, Biegel JA, Blaney S, et al.: Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24 (5): 337-42, 2002 Jun-Jul.
Geyer JR, Sposto R, Jennings M, et al.: Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children's Cancer Group. J Clin Oncol 23 (30): 7621-31, 2005.
Nicolaides T, Tihan T, Horn B, et al.: High-dose chemotherapy and autologous stem cell rescue for atypical teratoid/rhabdoid tumor of the central nervous system. J Neurooncol 98 (1): 117-23, 2010.
Gardner SL, Asgharzadeh S, Green A, et al.: Intensive induction chemotherapy followed by high dose chemotherapy with autologous hematopoietic progenitor cell rescue in young children newly diagnosed with central nervous system atypical teratoid rhabdoid tumors. Pediatr Blood Cancer 51 (2): 235-40, 2008.
Finkelstein-Shechter T, Gassas A, Mabbott D, et al.: Atypical teratoid or rhabdoid tumors: improved outcome with high-dose chemotherapy. J Pediatr Hematol Oncol 32 (5): e182-6, 2010.
Chi SN, Zimmerman MA, Yao X, et al.: Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27 (3): 385-9, 2009.
Reddy AT, Strother DR, Judkins AR, et al.: Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children's Oncology Group Trial ACNS0333. J Clin Oncol 38 (11): 1175-1185, 2020.
Zaky W, Dhall G, Ji L, et al.: Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: the Head Start III experience. Pediatr Blood Cancer 61 (1): 95-101, 2014.
Aridgides PD, Mahajan A, Eaton B, et al.: Focal versus craniospinal radiation for disseminated atypical teratoid/rhabdoid tumor following favorable response to systemic therapy. Pediatr Blood Cancer 70 (7): e30351, 2023.
Tekautz TM, Fuller CE, Blaney S, et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23 (7): 1491-9, 2005.
Buscariollo DL, Park HS, Roberts KB, et al.: Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118 (17): 4212-9, 2012.
Bartelheim K, Nemes K, Seeringer A, et al.: Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5 (8): 1765-75, 2016.
Frühwald MC, Hasselblatt M, Nemes K, et al.: Age and DNA methylation subgroup as potential independent risk factors for treatment stratification in children with atypical teratoid/rhabdoid tumors. Neuro Oncol 22 (7): 1006-1017, 2020.
Olson TA, Bayar E, Kosnik E, et al.: Successful treatment of disseminated central nervous system malignant rhabdoid tumor. J Pediatr Hematol Oncol 17 (1): 71-5, 1995.
Zimmerman MA, Goumnerova LC, Proctor M, et al.: Continuous remission of newly diagnosed and relapsed central nervous system atypical teratoid/rhabdoid tumor. J Neurooncol 72 (1): 77-84, 2005.
Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil recidivante
No hay un tratamiento estándar para niños con tumor teratoide rabdoide atípico (TTRA) del sistema nervioso central (SNC) recidivante.
Están en curso ensayos de terapia dirigida molecular. En un estudio del inhibidor EZH2 tazemetostat en adultos con sarcoma epitelioide y tumores rabdoides malignos fuera del sistema nervioso central (SNC) que exhibían pérdida de SMARCB1 o SMARCA4, se observó enfermedad estable prolongada, así como respuestas objetivas.[1] En el ensayo APEC1621C (NCT03213665) del Pediatric MATCH del Children's Oncology Group (COG) del Instituto Nacional del Cáncer (NCI), 8 niños con TTRA recibieron tazemetostat. Uno de los pacientes demostró estabilización de la enfermedad.[2][Nivel de evidencia B4]
La radioterapia estereotáctica o la radiocirugía estereotáctica, así como la radioterapia focal, también se pueden considerar para el tratamiento de niños con enfermedad recidivante.[3]
Los pacientes o familiares que deseen explorar otras terapias para la enfermedad deben contemplar la participación en ensayos con abordajes terapéuticos nuevos, porque no hay fármacos estándar que hayan demostrado actividad clínica significativa.
Independientemente de si se decide continuar con una terapia dirigida a la enfermedad en el momento de la progresión, el objetivo principal del tratamiento sigue siendo el cuidado paliativo. Esto garantiza que se aumenta al máximo la calidad de vida mientras se intenta reducir los síntomas y el estrés de la enfermedad terminal.
Opciones de tratamiento en evaluación clínica
Quizás haya ensayos clínicos terapéuticos de fase inicial para ciertos pacientes. Es posible que estos ensayos estén disponibles a través del Children's Oncology Group (COG), el Pediatric Brain Tumor Consortium u otras entidades. La información en inglés sobre los ensayos clínicos patrocinados por el Instituto Nacional del Cáncer (NCI) se encuentra en el portal de Internet del NCI. Para obtener información en inglés sobre ensayos clínicos patrocinados por otras organizaciones, consultar el portal de Internet ClinicalTrials.gov.
A continuación se presenta un ejemplo de ensayo clínico nacional o institucional en curso:
PEPN2121 (NCT05286801) (Tiragolumab and Atezolizumab for the Treatment of Relapsed or Refractory SMARCB1- or SMARCA4-Deficient Tumors): en este estudio se evalúa el uso de la combinación de un anticuerpo dirigido a PD-L1 (atezolizumab) con un anticuerpo dirigido a TIGIT (tiragolumab) para pacientes de tumores con deficiencia de SMARCB1 o SMARCA4. Los pacientes con TTRA podrían participar en este estudio.
Referencias:
Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.
Chi SN, Yi JS, Williams PM, et al.: Tazemetostat for tumors harboring SMARCB1/SMARCA4 or EZH2 alterations: results from NCI-COG pediatric MATCH APEC1621C. J Natl Cancer Inst 115 (11): 1355-1363, 2023.
Spina A, Gagliardi F, Boari N, et al.: Does Stereotactic Radiosurgery Positively Impact the Local Control of Atypical Teratoid Rhabdoid Tumors? World Neurosurg 104: 612-618, 2017.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Actualizaciones más recientes a este resumen (08 / 23 / 2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes incorporados en este resumen a partir de la fecha arriba indicada.
Este resumen fue objeto de revisión integral.
El Consejo editorial del PDQ sobre el tratamiento pediátrico es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento pediátrico, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del tumor teratoide rabdoide atípico del sistema nervioso central infantil son:
Kenneth J. Cohen, MD, MBA (Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins Hospital)
Karen J. Marcus, MD, FACR (Dana-Farber Cancer Institute/Boston Children's Hospital)
Roger J. Packer, MD (Children's National Hospital)
D. Williams Parsons, MD, PhD (Texas Children's Hospital)
Malcolm A. Smith, MD, PhD (National Cancer Institute)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento pediátrico emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.