Esta información es producida y suministrada por el Instituto Nacional del Cáncer (NCI, por sus siglas en inglés). La información en este tema puede haber cambiado desde que se escribió. Para la información más actual, comuníquese con el Instituto Nacional del Cáncer a través del Internet en la página web http://cancer.gov o llame al 1-800-4-CANCER.
Información general sobre el linfoma no Hodgkin periférico de células T
Los linfomas no Hodgkin (LNH o linfomas no hodgkinianos) de células T son un grupo heterogéneo de neoplasias malignas linfoproliferativas de células o linfocitos T, que representan menos del 15 % de los LNH.[1] Alrededor del 85 % de los casos de LNH son linfomas de células B. Para obtener más información, consultar Tratamiento del linfoma no Hodgkin de células B.
Los linfomas de células T se dividen en linfomas cutáneos de células T, linfomas no Hodgkin periféricos de células T y el linfoma linfoblástico de células T o leucemia linfocítica aguda.
El linfoma linfoblástico de células T o leucemia linfoblástica aguda es una enfermedad que surge de células T muy tempranas, suele comprometer el timo y es más común en adultos jóvenes. A menudo, la enfermedad del tipo linfoma se trata de manera similar a la del tipo leucemia. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda.
Los linfomas cutáneos de células T comienzan en la piel e incluyen, entre otros, la micosis fungoide, el síndrome de Sézary y el linfoma anaplásico de células grandes cutáneo primario. Para obtener más información, consultar Tratamiento de la micosis fungoide y otros linfomas cutáneos de células T.
Los linfomas no Hodgkin periféricos de células T se originan a partir de células T maduras. También se llaman linfomas periféricos de células T y LPCT. Por lo general, este tipo de linfomas surge en los tejidos linfoides y a veces se disemina a otros órganos. Los subconjuntos de LPCT incluyen el linfoma anaplásico de células grandes (LACG), el linfoma angioinmunoblástico de células T (LACT), el linfoma extraganglionar de células T-NK, los LPCT sin otra indicación (LPCT-SAI), la leucemia o linfoma de células T en adultos (LLCTA), el linfoma de células T asociado a enteropatía (LTAE), el linfoma hepatoesplénico de células T, la leucemia prolinfocítica de células T (LPL-T) y otros.
Incidencia y mortalidad
Los linfomas de células T representan menos del 15 % de los casos de LNH. La mayoría de los subtipos de linfoma de células T se relacionan con desenlaces más precarios que los de los linfomas de células B.[1]
Características anatómicas
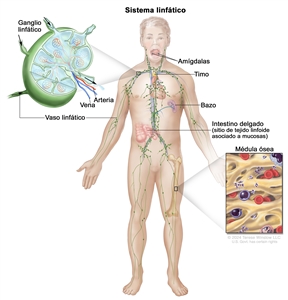
El LNH por lo general se origina en los tejidos linfoides.
Pronóstico y supervivencia
El pronóstico del LPCT varía según el subtipo, el estadio y otros factores. En general, el LPCT se relaciona con un pronóstico precario, con una tasa de supervivencia a 5 años del 30 % al 40 %.[2,3] Si bien los desenlaces son mejores para los pacientes con LACG positivo para ALK, con una mediana de supervivencia general (SG) a 5 años del 70 % al 80 %,[2,3,4] otros subconjuntos se relacionan con una supervivencia más precaria, como el LACG negativo para ALK, el LACT, el LPCT-SAI, el linfoma hepatoesplénico de células T, el LTAE, y el linfoma extraganglionar de células T-NK.[5,6]
A diferencia de los LNH de células B, que incluyen tipos de linfoma de crecimiento lento (es decir, de bajo grado de malignidad o indolente) y de crecimiento rápido (es decir, de alto grado de malignidad o agresivos), la mayoría de los LPCT se consideran de crecimiento rápido.[7] Al igual que con la mayoría de los otros linfomas de crecimiento rápido, los LPCT a menudo se curan con terapia sistémica, aunque las opciones de tratamiento eficaces son más limitadas, en particular en el entorno de recaída o resistencia al tratamiento.[8,9]
Aunque los tratamientos existentes son curativos en una proporción significativa de los pacientes con linfoma, se están llevando a cabo numerosos ensayos clínicos para mejorar el tratamiento. En lo posible, los pacientes se deben incluir en estos estudios.
Además de los exámenes de detección del VIH en pacientes con linfomas de crecimiento rápido, se puede evaluar la presencia de hepatitis B o C activa antes del tratamiento con quimioterapia.[10,11] Los pacientes con infección por el virus de la hepatitis B (VHB) detectable se benefician de la profilaxis con entecavir.[12,13] Los pacientes con infección por el VHB resuelta (es decir, negativa para el antígeno de superficie del virus de la hepatitis B, pero positiva para el anticuerpo central del virus de la hepatitis B), presentan riesgo de reactivación de la infección por el VHB y necesitan vigilancia del DNA del VHB. La terapia profiláctica con nucleósidos redujo la reactivación de la infección por el VHB de un 10,8 % a un 2,1 % en un estudio retrospectivo de 326 pacientes.[14] Los pacientes que reciben quimioterapia combinada suelen recibir profilaxis con aciclovir o valaciclovir para el virus del herpes zóster y profilaxis con trimetoprima-sulfametoxazol o dapsona para la infección por Pneumocystis.
Referencias:
American Cancer Society: Types of T-cell lymphoma. American Cancer Society, 2018. Available online. Last accessed April 5, 2024.
Vose J, Armitage J, Weisenburger D, et al.: International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol 26 (25): 4124-30, 2008.
Ellin F, Landström J, Jerkeman M, et al.: Real-world data on prognostic factors and treatment in peripheral T-cell lymphomas: a study from the Swedish Lymphoma Registry. Blood 124 (10): 1570-7, 2014.
Sibon D, Fournier M, Brière J, et al.: Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol 30 (32): 3939-46, 2012.
Petrich AM, Helenowski IB, Bryan LJ, et al.: Factors predicting survival in peripheral T-cell lymphoma in the USA: a population-based analysis of 8802 patients in the modern era. Br J Haematol 168 (5): 708-18, 2015.
Foss FM, Horwitz SM, Civallero M, et al.: Incidence and outcomes of rare T cell lymphomas from the T Cell Project: hepatosplenic, enteropathy associated and peripheral gamma delta T cell lymphomas. Am J Hematol 95 (2): 151-155, 2020.
Armitage JO: The aggressive peripheral T-cell lymphomas: 2017. Am J Hematol 92 (7): 706-715, 2017.
Bellei M, Foss FM, Shustov AR, et al.: The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, International T-Cell Project. Haematologica 103 (7): 1191-1197, 2018.
Lansigan F, Horwitz SM, Pinter-Brown LC, et al.: Outcomes for Relapsed and Refractory Peripheral T-Cell Lymphoma Patients after Front-Line Therapy from the COMPLETE Registry. Acta Haematol 143 (1): 40-50, 2020.
Niitsu N, Hagiwara Y, Tanae K, et al.: Prospective analysis of hepatitis B virus reactivation in patients with diffuse large B-cell lymphoma after rituximab combination chemotherapy. J Clin Oncol 28 (34): 5097-100, 2010.
Dong HJ, Ni LN, Sheng GF, et al.: Risk of hepatitis B virus (HBV) reactivation in non-Hodgkin lymphoma patients receiving rituximab-chemotherapy: a meta-analysis. J Clin Virol 57 (3): 209-14, 2013.
Huang YH, Hsiao LT, Hong YC, et al.: Randomized controlled trial of entecavir prophylaxis for rituximab-associated hepatitis B virus reactivation in patients with lymphoma and resolved hepatitis B. J Clin Oncol 31 (22): 2765-72, 2013.
Li H, Zhang HM, Chen LF, et al.: Prophylactic lamivudine to improve the outcome of HBsAg-positive lymphoma patients during chemotherapy: a systematic review and meta-analysis. Clin Res Hepatol Gastroenterol 39 (1): 80-92, 2015.
Kusumoto S, Arcaini L, Hong X, et al.: Risk of HBV reactivation in patients with B-cell lymphomas receiving obinutuzumab or rituximab immunochemotherapy. Blood 133 (2): 137-146, 2019.
Efectos tardíos del tratamiento del linfoma no Hodgkin periférico de células T
Se han observado efectos tardíos del tratamiento del linfoma no Hodgkin (LNH). Es posible que se presente una alteración de la fertilidad o la capacidad reproductiva después de la exposición a alquilantes.[1] Hasta tres décadas después del diagnóstico, los pacientes tienen un riesgo significativamente elevado de presentar segundos cánceres primarios, en especial los siguientes:[2,3,4,5]
Cáncer de pulmón.
Cáncer de encéfalo.
Cáncer de riñón.
Cáncer de vejiga.
Melanoma.
Linfoma de Hodgkin.
Leucemia no linfocítica aguda.
La disfunción ventricular izquierda fue un efecto tardío significativo en los sobrevivientes a largo plazo de LNH de grado alto que recibieron más de 200 mg/m² de doxorrubicina.[1,6]
El síndrome mielodisplásico y la leucemia mielógena aguda son complicaciones tardías de la terapia mielosupresora con apoyo de médula ósea autógena o células madre periféricas, así como de la quimioterapia convencional con alquilantes.[3,7,8,9,10,11,12,13,14] La mayoría de estos pacientes presentan hematopoyesis clonal incluso antes del trasplante, lo que indica que la lesión hematológica por lo general se presenta durante la quimioterapia de inducción o reinducción.[9,15,16] Se realizó un seguimiento, durante una mediana de 10 años, de una serie de 605 pacientes que recibieron trasplante de médula ósea (TMO) autógeno con ciclofosfamida y radioterapia corporal total (como acondicionamiento). La incidencia de una segunda neoplasia maligna fue del 21 %, y el 10 % de esas neoplasias malignas fueron tumores sólidos.[17]
En un estudio de mujeres jóvenes que recibieron TMO autógeno, se notificaron embarazos exitosos con bebés que nacieron sin anomalías congénitas.[18] La tromboembolia venosa tardía se puede presentar después de un TMO alogénico o autógeno.[19]
Algunos pacientes presentan osteopenia u osteoporosis al inicio del tratamiento; es posible que la densidad ósea empeore después del tratamiento para el linfoma.[20]
Referencias:
Haddy TB, Adde MA, McCalla J, et al.: Late effects in long-term survivors of high-grade non-Hodgkin's lymphomas. J Clin Oncol 16 (6): 2070-9, 1998.
Travis LB, Curtis RE, Glimelius B, et al.: Second cancers among long-term survivors of non-Hodgkin's lymphoma. J Natl Cancer Inst 85 (23): 1932-7, 1993.
Mudie NY, Swerdlow AJ, Higgins CD, et al.: Risk of second malignancy after non-Hodgkin's lymphoma: a British Cohort Study. J Clin Oncol 24 (10): 1568-74, 2006.
Hemminki K, Lenner P, Sundquist J, et al.: Risk of subsequent solid tumors after non-Hodgkin's lymphoma: effect of diagnostic age and time since diagnosis. J Clin Oncol 26 (11): 1850-7, 2008.
Major A, Smith DE, Ghosh D, et al.: Risk and subtypes of secondary primary malignancies in diffuse large B-cell lymphoma survivors change over time based on stage at diagnosis. Cancer 126 (1): 189-201, 2020.
Moser EC, Noordijk EM, van Leeuwen FE, et al.: Long-term risk of cardiovascular disease after treatment for aggressive non-Hodgkin lymphoma. Blood 107 (7): 2912-9, 2006.
Darrington DL, Vose JM, Anderson JR, et al.: Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol 12 (12): 2527-34, 1994.
Stone RM, Neuberg D, Soiffer R, et al.: Myelodysplastic syndrome as a late complication following autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 12 (12): 2535-42, 1994.
Armitage JO, Carbone PP, Connors JM, et al.: Treatment-related myelodysplasia and acute leukemia in non-Hodgkin's lymphoma patients. J Clin Oncol 21 (5): 897-906, 2003.
André M, Mounier N, Leleu X, et al.: Second cancers and late toxicities after treatment of aggressive non-Hodgkin lymphoma with the ACVBP regimen: a GELA cohort study on 2837 patients. Blood 103 (4): 1222-8, 2004.
Oddou S, Vey N, Viens P, et al.: Second neoplasms following high-dose chemotherapy and autologous stem cell transplantation for malignant lymphomas: a report of six cases in a cohort of 171 patients from a single institution. Leuk Lymphoma 31 (1-2): 187-94, 1998.
Lenz G, Dreyling M, Schiegnitz E, et al.: Moderate increase of secondary hematologic malignancies after myeloablative radiochemotherapy and autologous stem-cell transplantation in patients with indolent lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group. J Clin Oncol 22 (24): 4926-33, 2004.
McLaughlin P, Estey E, Glassman A, et al.: Myelodysplasia and acute myeloid leukemia following therapy for indolent lymphoma with fludarabine, mitoxantrone, and dexamethasone (FND) plus rituximab and interferon alpha. Blood 105 (12): 4573-5, 2005.
Morton LM, Curtis RE, Linet MS, et al.: Second malignancy risks after non-Hodgkin's lymphoma and chronic lymphocytic leukemia: differences by lymphoma subtype. J Clin Oncol 28 (33): 4935-44, 2010.
Mach-Pascual S, Legare RD, Lu D, et al.: Predictive value of clonality assays in patients with non-Hodgkin's lymphoma undergoing autologous bone marrow transplant: a single institution study. Blood 91 (12): 4496-503, 1998.
Lillington DM, Micallef IN, Carpenter E, et al.: Detection of chromosome abnormalities pre-high-dose treatment in patients developing therapy-related myelodysplasia and secondary acute myelogenous leukemia after treatment for non-Hodgkin's lymphoma. J Clin Oncol 19 (9): 2472-81, 2001.
Brown JR, Yeckes H, Friedberg JW, et al.: Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin's lymphoma. J Clin Oncol 23 (10): 2208-14, 2005.
Jackson GH, Wood A, Taylor PR, et al.: Early high dose chemotherapy intensification with autologous bone marrow transplantation in lymphoma associated with retention of fertility and normal pregnancies in females. Scotland and Newcastle Lymphoma Group, UK. Leuk Lymphoma 28 (1-2): 127-32, 1997.
Gangaraju R, Chen Y, Hageman L, et al.: Risk of venous thromboembolism in patients with non-Hodgkin lymphoma surviving blood or marrow transplantation. Cancer 125 (24): 4498-4508, 2019.
Westin JR, Thompson MA, Cataldo VD, et al.: Zoledronic acid for prevention of bone loss in patients receiving primary therapy for lymphomas: a prospective, randomized controlled phase III trial. Clin Lymphoma Myeloma Leuk 13 (2): 99-105, 2013.
Clasificación celular del linfoma no Hodgkin periférico de células T
Se debe consultar con un patólogo antes de realizar una biopsia porque algunos estudios exigen una preparación especial del tejido (por ejemplo, tejido congelado). El conocimiento de los marcadores de superficie celular y los reordenamientos de los genes de inmunoglobulinas y del receptor de células T puede ser útil al momento de tomar decisiones diagnósticas y terapéuticas. El exceso clonal de cadenas ligeras de inmunoglobulina permite diferenciar las células linfoides malignas de las células reactivas. Es muy importante que un hematopatólogo con experiencia en el diagnóstico de linfomas realice un examen minucioso de las muestras de biopsia externas porque las características histopatológicas afectan el pronóstico y el abordaje del tratamiento. Aunque se recomienda obtener biopsias de ganglios linfáticos cuando sea posible, a veces los datos inmunofenotípicos son suficientes para establecer un diagnóstico de linfoma cuando es preferible hacer un estudio citológico mediante aspiración con aguja fina o biopsia con aguja gruesa.[1,2]
Sistemas de clasificación actuales
Clasificación REAL modificada por la Organización Mundial de la Salud
En la modificación que la Organización Mundial de la Salud (OMS) hizo a la clasificación europea americana revisada del linfoma (Revised European American Lymphoma [REAL]) se reconocen tres categorías principales de neoplasias linfoides malignas según la morfología y el linaje celular: neoplasias de células B, neoplasias de células T o de células citolíticas naturales (NK), y linfoma de Hodgkin (LH). En esta clasificación, se incluyen los linfomas y las leucemias linfoides porque las fases sólidas y circulantes de estas enfermedades se encuentran en muchas neoplasias linfoides, así que la distinción entre ellas es artificial. Por ejemplo, la leucemia linfocítica crónica (LLC) de células B y el linfoma linfocítico de células B pequeñas son distintas manifestaciones de la misma neoplasia, al igual que los linfomas linfoblásticos y las leucemias linfocíticas agudas. Dentro de las categorías de células B y células T, se reconocen dos subdivisiones: neoplasias precursoras, que corresponden a los estadios más tempranos de diferenciación, y neoplasias diferenciadas más maduras.[3,4]
Neoplasias de células B
Neoplasias de células B precursoras: leucemia linfoblástica aguda de células B precursoras o linfoma linfoblástico de células B precursoras.
Neoplasias de células B periféricas.
Leucemia linfocítica crónica de células B o linfoma linfocítico de células B pequeñas.
Leucemia prolinfocítica de células B.
Linfoma linfoplasmocítico o inmunocitoma.
Linfoma de células de manto.
Linfoma folicular.
Linfoma extraganglionar de células B de zona marginal de tejido linfoide asociado a mucosas.
Linfoma ganglionar de células B de zona marginal (± células B monocitoides).
Linfoma esplénico de zona marginal (± linfocitos vellosos).
Leucemia de células pilosas.
Plasmocitoma o mieloma de células plasmáticas.
Linfoma difuso de células B grandes.
Linfoma de Burkitt.
Neoplasias de células T y posiblemente derivadas de células NK
Neoplasias de células T precursoras: leucemia linfoblástica aguda de células T precursoras o linfoma linfoblástico de células T precursoras. Para obtener más información, consultar Tratamiento de la leucemia linfoblástica aguda en adultos.
Neoplasias periféricas de células T-NK.
Leucemia linfocítica crónica de células T o leucemia prolinfocítica de células T.
Leucemia linfocítica granular de células T.
Micosis fungoide (incluso el síndrome de Sézary).
Linfoma periférico de células T sin otra indicación.
Linfoma hepatoesplénico de células T γ-δ.
Linfoma subcutáneo de células T similar a la paniculitis.
Linfoma angioinmunoblástico de células T.
Linfoma extraganglionar de células T-NK de tipo nasal.
Linfoma de células T asociado a enteropatía.
Leucemia o linfoma de células T en adultos (virus linfotrópico humano de células T de tipo 1 [VLHT] 1+).
Linfoma anaplásico de células grandes de tipo sistémico primario.
Linfoma anaplásico de células grandes de tipo cutáneo primario.
Leucemia de células NK de crecimiento rápido (agresiva).
Linfoma de Hodgkin
Linfoma de Hodgkin con predominio linfocítico nodular.
Linfoma de Hodgkin clásico.
Linfoma de Hodgkin con esclerosis nodular.
Linfoma de Hodgkin clásico rico en linfocitos.
Linfoma de Hodgkin con celularidad mixta.
Linfoma de Hodgkin con agotamiento linfocítico.
La clasificación REAL abarca todas las neoplasias linfoproliferativas. Para obtener más información, consultar los siguientes resúmenes del PDQ:
Tratamiento de la leucemia linfoblástica aguda en adultos
Tratamiento del linfoma de Hodgkin
Tratamiento del linfoma relacionado con el SIDA
Tratamiento de la leucemia linfocítica crónica
Tratamiento de la leucemia de células pilosas
Tratamiento de la micosis fungoide y otros linfomas cutáneos de células T
Tratamiento de las neoplasias de células plasmáticas (incluso mieloma múltiple)
Tratamiento del linfoma primario del sistema nervioso central
Subtipos del linfoma no Hodgkin periférico de células T
Los linfomas no Hodgkin periféricos de células T incluyen, entre otros, los siguientes subtipos:
Linfoma anaplásico de células grandes.
Linfoma angioinmunoblástico de células T.
Linfoma periférico de células T sin otra indicación.
Linfoma extraganglionar de células T-NK.
Linfoma de células T asociado a enteropatía.
Linfoma hepatoesplénico de células T.
Leucemia o linfoma de células T en adultos.
Leucemia prolinfocítica de células T. Para obtener más información, consultar Tratamiento de la leucemia linfocítica crónica.
Linfomas cutáneos de células T, como la micosis fungoide y el síndrome de Sézary, el linfoma subcutáneo de células T similar a la paniculitis, el linfoma anaplásico de células grandes cutáneo primario, el linfoma cutáneo primario de células T γ-δ, y otros). Para obtener más información, consultar Tratamiento de la micosis fungoide (incluso el síndrome de Sézary).
Leucemia linfocítica granular de células T. Para obtener más información, consultar Tratamiento de la leucemia linfocítica crónica.
Referencias:
Zeppa P, Marino G, Troncone G, et al.: Fine-needle cytology and flow cytometry immunophenotyping and subclassification of non-Hodgkin lymphoma: a critical review of 307 cases with technical suggestions. Cancer 102 (1): 55-65, 2004.
Young NA, Al-Saleem T: Diagnosis of lymphoma by fine-needle aspiration cytology using the revised European-American classification of lymphoid neoplasms. Cancer 87 (6): 325-45, 1999.
Pileri SA, Milani M, Fraternali-Orcioni G, et al.: From the R.E.A.L. Classification to the upcoming WHO scheme: a step toward universal categorization of lymphoma entities? Ann Oncol 9 (6): 607-12, 1998.
Society for Hematopathology Program: Society for Hematopathology Program. Am J Surg Pathol 21 (1): 114-121, 1997.
Información sobre los estadios del linfoma no Hodgkin periférico de células T
El estadio es importante para la selección del tratamiento en pacientes con linfoma no Hodgkin (LNH). Por lo general, las tomografías computarizadas (TC) del tórax y el abdomen forman parte de la evaluación para la estadificación de todos los pacientes con linfoma. El sistema de estadificación para el LNH es similar al que se usa para el linfoma de Hodgkin (LH).
Es común que los pacientes con LNH presenten compromiso de los siguientes sitios:
Ganglios linfáticos no adyacentes.
Anillo de Waldeyer.
Ganglios epitrocleares.
Tubo digestivo.
Sitios extraganglionares. (En ocasiones, una localización extraganglionar es el único sitio comprometido en pacientes con un linfoma difuso).
Médula ósea.
Hígado (muy común en pacientes con linfomas de grado bajo).
El examen citológico del líquido cefalorraquídeo a veces da un resultado positivo en pacientes con LNH de crecimiento rápido. El compromiso de los ganglios linfáticos hiliares y mediastínicos es menos común que en los pacientes con LH. Sin embargo, las adenopatías mediastínicas son una característica destacada del linfoma linfoblástico y del linfoma mediastínico primario de células B, que se presentan sobre todo en adultos jóvenes.
La mayoría de los pacientes con LNH consultan con una enfermedad avanzada (estadio III o IV) que a menudo se identifica mediante tomografías computarizadas (TC) o biopsias de la médula ósea y de otros sitios comprometidos accesibles. En una revisión retrospectiva de más de 32 000 casos de linfoma en Francia, hasta el 40 % de los diagnósticos se confirmaron mediante biopsia con aguja gruesa y el 60 % mediante biopsia por escisión.[1] Después de la revisión por expertos, la biopsia con aguja gruesa proporcionó un diagnóstico definitivo en el 92,3 % de los casos; mientras que la biopsia por escisión proporcionó un diagnóstico definitivo en el 98,1 % de los casos (P < 0,0001). Por lo general, no se requiere una biopsia laparoscópica o laparotomía para la estadificación, pero es posible que en raras ocasiones se necesite para establecer un diagnóstico o determinar el tipo histológico del linfoma.[2]
La tomografía por emisión de positrones (TEP) con flúor F 18-fludesoxiglucosa se puede usar para la estadificación inicial. También quizás se utilice durante el seguimiento después del tratamiento como complemento de la TC.[3] En múltiples estudios se demostró que las TEP intermedias después de 2 a 4 ciclos de terapia no aportan información pronóstica confiable. En un ensayo de grupo cooperativo grande (ECOG-E344 [NCT00274924]) se notificaron problemas de reproducibilidad entre los observadores. En 2 ensayos prospectivos y en 1 metanálisis no se observaron diferencias en los desenlaces entre los pacientes con resultados negativos en la TEP y los pacientes con resultados positivos en la TEP pero que tenían resultados negativos en la biopsia.[4,5,6,7]
En un estudio retrospectivo de 130 pacientes con linfoma difuso de células B grandes, la TEP permitió identificar el compromiso de la médula ósea con importancia clínica; la biopsia de médula ósea no llevó a la sobreestadificación de ningún paciente con linfoma.[8] En un estudio retrospectivo de 580 pacientes con linfoma folicular de 7 ensayos patrocinados por el Instituto Nacional del Cáncer, no se observó ninguna mejora en la evaluación de la respuesta al tratamiento cuando se añadió la biopsia de la médula ósea a las imágenes radiológicas.[9] La evaluación del LNH debe incluir una biopsia de la médula ósea cuando el resultado posiblemente conlleve un cambio en el tratamiento (por ejemplo, si se encuentra un estadio limitado vs. uno avanzado) o durante la evaluación de la presencia de citopenias.
Sistema de estadificación
Clasificación de Lugano
El American Joint Committee on Cancer (AJCC) adoptó la clasificación de Lugano para evaluar y estadificar el linfoma.[10] El sistema de clasificación de Lugano reemplazó el sistema de clasificación de Ann Arbor, que se adoptó en 1971 en la conferencia de Ann Arbor,[11] y que fue modificado 18 años después en la reunión de Cotswolds.[12,13]
Cuadro 1. Clasificación de Lugano para el linfoma de Hodgkin y el linfoma no Hodgkina
Estadio
Descripción del estadio
Imagen
LCR = líquido cefalorraquídeo; TC = tomografía computarizada; LDCBG = linfoma difuso de células B grandes; LNH = linfoma no Hodgkin.
a Hodgkin and Non-Hodgkin Lymphomas. En: Amin MB, Edge SB, Greene FL, et al., eds.:AJCC Cancer Staging Manual. 8.ª edición Nueva York, NY: Springer, 2017, pp. 937-58.
b El estadio II con gran masa tumoral (tumor voluminoso) quizás se considere un estadio temprano o avanzado en función de las características histológicas y los factores pronósticos del linfoma.
c La definición de una gran masa tumoral varía según las características histológicas del linfoma. En la clasificación de Lugano, una gran masa tumoral en el linfoma de Hodgkin se define como una masa que mide más de un tercio del diámetro torácico en la TC del tórax, o una masa que mide >10 cm. Las definiciones recomendadas para una gran masa tumoral en el LNH varían de acuerdo a las características histológicas del linfoma. En el linfoma folicular, se sugirió la medida de 6 cm a partir del Follicular Lymphoma International Prognostic Index-2 y su validación. En el LDCBG, se han usado valores de corte que oscilan entre 5 cm y 10 cm, aunque el límite recomendado es de 10 cm.
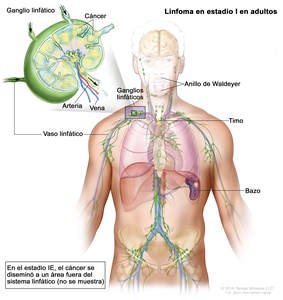
Estadio limitado
I
Compromiso de un solo sitio linfático (es decir, una región ganglionar, el anillo de Waldeyer, el timo o el bazo).
IE
Compromiso de 1 solo sitio extralinfático sin compromiso ganglionar (infrecuente en el linfoma de Hodgkin).
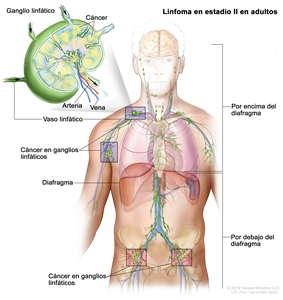
II
Compromiso de 2 o más regiones ganglionares en el mismo lado del diafragma.
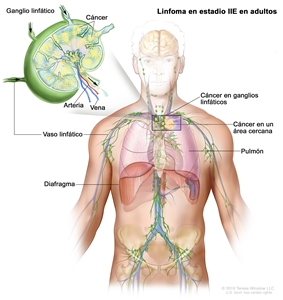
IIE
Diseminación extralinfática adyacente desde un sitio ganglionar con compromiso de otras regiones ganglionares en el mismo lado del diafragma o sin este.
II con gran masa tumoralb
Estadio II con gran masa tumoralc
Estadio avanzado
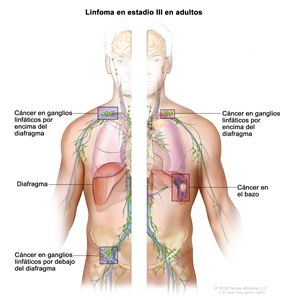
III
Compromiso de regiones ganglionares en ambos lados del diafragma; adenopatías por encima del diafragma y compromiso esplénico.
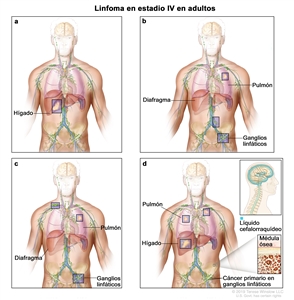
IV
Compromiso difuso o diseminación a uno o más órganos extralinfáticos con compromiso ganglionar o sin este; compromiso de órgano extralinfático no adyacente con enfermedad ganglionar en estadio II; o compromiso de cualquier órgano extralinfático con enfermedad ganglionar en estadio III. El estadio IV incluye cualquier tipo de compromiso del LCR, la médula ósea y el hígado, o la presencia de lesiones pulmonares múltiples (diferentes a las lesiones por diseminación directa de una enfermedad en estadio IIE).
Nota: Se utiliza la designación A o B junto con el grupo de estadio para el linfoma de Hodgkin. En el LNH ya no se utiliza la designación A ni B.
En ocasiones, se usan otros sistemas de estadificación especializados. El médico debe estar al tanto del sistema que se usa en el informe del paciente.
La designación E se usa cuando aparecen neoplasias linfoides extraganglionares malignas en tejidos separados de los conglomerados linfáticos principales pero cercanos a estos. El estadio IV indica una enfermedad con diseminación difusa por todo un sitio extraganglionar, como el hígado. Si el compromiso de uno o más sitios extralinfáticos se documentó mediante estudio patológico, se usa el símbolo del sitio comprometido seguido por el signo más (+).
Cuadro 2. Notación para la identificación de sitios específicos
N = ganglios
H = hígado
L = pulmón
M = médula ósea
S = bazo
P = pleura
O = hueso
D = piel
En la práctica actual se asigna un estadio clínico a partir de los hallazgos de la evaluación clínica y un estadio patológico a partir de los hallazgos de los procedimientos invasivos adicionales a la biopsia inicial.
Por ejemplo, es posible que se encuentre un compromiso del hígado y la médula ósea mediante una biopsia percutánea en un paciente con adenopatía inguinal sin síntomas sistémicos que tiene un resultado positivo en el linfangiograma. El estadio exacto para dicho paciente sería estadio clínico IIA, estadio patológico IVA(H+)(M+).
Hay otros factores que no se incluyen en el sistema de estadificación anterior, pero que son importantes para la estadificación y el pronóstico de los pacientes con LNH. Estos factores son los siguientes:
Edad.
Estado funcional (EF).
Tamaño del tumor.
Concentraciones de lactato–deshidrogenasa (LDH).
Número de sitios con compromiso extraganglionar.
En el índice pronóstico internacional de la National Comprehensive Cancer Network (National Comprehensive Cancer Network International Prognostic Index [NCCN-IPI]) para el linfoma no Hodgkin (LNH) de crecimiento rápido (linfoma difuso de células grandes) se identifican los siguientes 5 factores de riesgo significativos para el pronóstico de la supervivencia general (SG) y los puntajes de riesgo relacionado:[14]
Edad.
<40 años: 0.
41–60 años: 1.
61–75 años: 2.
>75 años: 3.
Estadio III o IV: 1.
Estado funcional (EF) 2, 3 o 4: 1.
Concentración sérica de lactato–deshidrogenasa (LDH).
Normalizada: 0.
>1x–3x: 1.
>3x: 2.
Número de sitios extraganglionares ≥2: 1.
Puntajes de riesgo:
Bajo (0 o 1): Tasa de SG a 5 años, 96 %; tasa de supervivencia sin progresión (SSP), 91 %.
Intermedio bajo (2 o 3): Tasa de SG a 5 años, 82 %; tasa de SSP, 74 %.
Intermedio alto (4 o 5): Tasa de SG a 5 años, 64 %; tasa de SSP, 51 %.
Alto (>6): Tasa de SG a 5 años, 33 %; tasa de SSP, 30 %.
Referencias:
Syrykh C, Chaouat C, Poullot E, et al.: Lymph node excisions provide more precise lymphoma diagnoses than core biopsies: a French Lymphopath network survey. Blood 140 (24): 2573-2583, 2022.
Mann GB, Conlon KC, LaQuaglia M, et al.: Emerging role of laparoscopy in the diagnosis of lymphoma. J Clin Oncol 16 (5): 1909-15, 1998.
Barrington SF, Mikhaeel NG, Kostakoglu L, et al.: Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 32 (27): 3048-58, 2014.
Horning SJ, Juweid ME, Schöder H, et al.: Interim positron emission tomography scans in diffuse large B-cell lymphoma: an independent expert nuclear medicine evaluation of the Eastern Cooperative Oncology Group E3404 study. Blood 115 (4): 775-7; quiz 918, 2010.
Moskowitz CH, Schöder H, Teruya-Feldstein J, et al.: Risk-adapted dose-dense immunochemotherapy determined by interim FDG-PET in Advanced-stage diffuse large B-Cell lymphoma. J Clin Oncol 28 (11): 1896-903, 2010.
Pregno P, Chiappella A, Bellò M, et al.: Interim 18-FDG-PET/CT failed to predict the outcome in diffuse large B-cell lymphoma patients treated at the diagnosis with rituximab-CHOP. Blood 119 (9): 2066-73, 2012.
Sun N, Zhao J, Qiao W, et al.: Predictive value of interim PET/CT in DLBCL treated with R-CHOP: meta-analysis. Biomed Res Int 2015: 648572, 2015.
Khan AB, Barrington SF, Mikhaeel NG, et al.: PET-CT staging of DLBCL accurately identifies and provides new insight into the clinical significance of bone marrow involvement. Blood 122 (1): 61-7, 2013.
Rutherford SC, Yin J, Pederson L, et al.: Relevance of Bone Marrow Biopsies for Response Assessment in US National Cancer Institute National Clinical Trials Network Follicular Lymphoma Clinical Trials. J Clin Oncol 41 (2): 336-342, 2023.
Hodgkin and non-Hodgkin lymphoma. In: Amin MB, Edge SB, Greene FL, et al., eds.: AJCC Cancer Staging Manual. 8th ed. Springer; 2017, pp. 937–58.
Carbone PP, Kaplan HS, Musshoff K, et al.: Report of the Committee on Hodgkin's Disease Staging Classification. Cancer Res 31 (11): 1860-1, 1971.
Lister TA, Crowther D, Sutcliffe SB, et al.: Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin's disease: Cotswolds meeting. J Clin Oncol 7 (11): 1630-6, 1989.
National Cancer Institute sponsored study of classifications of non-Hodgkin's lymphomas: summary and description of a working formulation for clinical usage. The Non-Hodgkin's Lymphoma Pathologic Classification Project. Cancer 49 (10): 2112-35, 1982.
Zhou Z, Sehn LH, Rademaker AW, et al.: An enhanced International Prognostic Index (NCCN-IPI) for patients with diffuse large B-cell lymphoma treated in the rituximab era. Blood 123 (6): 837-42, 2014.
Tratamiento del linfoma anaplásico de células grandes
El linfoma anaplásico de células grandes (LACG) es un linfoma periférico de células T relacionado con el antígeno CD30. La translocación de los cromosomas 2 y 5 crea una proteína de fusión única de nucleofosmina y la cinasa del linfoma anaplásico (ALK).[1,2] Los pacientes cuyos linfomas expresan ALK en pruebas inmunohistoquímicas son por lo general más jóvenes y a veces presentan síntomas sistémicos, compromiso extraganglionar y enfermedad en estadio avanzado. Sin embargo, tienen una tasa de supervivencia más favorable que los pacientes con enfermedad negativa para ALK.[3,4]
En un ensayo aleatorizado prospectivo se incluyeron 452 pacientes con linfoma de células T positivo para CD30 (expresión de CD30 >10 %). De estos pacientes, el 70 % tenían LACG (22 % con enfermedad positiva para ALK y 48 % con enfermedad negativa para ALK). En el ensayo se comparó el régimen estándar utilizado antes, CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona), con brentuximab vedotina (un anticuerpo monoclonal anti-CD30 conjugado con un citotóxico) combinado con ciclofosfamida, doxorrubicina y prednisona (régimen A+CHP).[5]
Después de una mediana de seguimiento de 47,6 meses, las tasas de supervivencia general (SG) a 5 años fueron del 70,1 % (intervalo de confianza [IC] 95 %, 63,3–75,9 %) para los pacientes que recibieron A+CHP y del 61,0 % (IC 95 %, 54,0–67,3 %) para los pacientes que recibieron CHOP (cociente de riesgos instantáneos [CRI], 0,72; IC 95 %, 0,53–0,99).[6][Nivel de evidencia A1]
Las tasas de supervivencia sin progresión (SSP) a 5 años fueron del 51,4 % (IC 95 %, 42,8–59,4 %) para los pacientes que recibieron A+CHP y del 43,0 % (IC 95 %, 35,8–50,0 %) para los pacientes que recibieron CHOP (CRI, 0,70; IC 95 %, 0,53–0,91).
Esto permitió establecer el régimen A+CHP como una nueva opción para los pacientes con LACG u otros linfomas de células T positivos para CD30, como el linfoma angioinmunoblástico de células T y el linfoma periférico de células T sin otra indicación.
En pacientes con recaída de la enfermedad, se notificaron respuestas anecdóticas con brentuximab vedotina,[7,8,9,10] romidepsina[11] y pralatrexato.[12][Nivel de evidencia C3]
En un estudio de fase II (NCT00866047), el 66 % de 58 pacientes lograron una respuesta completa con el brentuximab vedotina.[10]
Al cabo de una mediana de seguimiento de 58 meses, la tasa de SSP a 5 años fue del 57 % (IC 95 %, 41–74 %) y la tasa de SG a 5 años fue del 79 % (IC 95 %, 65–92 %). De los pacientes que lograron una respuesta completa, el 42 % se sometieron a trasplante de células madre (TCM) hematopoyéticas.[10][Nivel de evidencia C3]
En una revisión retrospectiva, 39 pacientes con recaída de la enfermedad tuvieron una tasa de SSP a 3 años del 50 % después de un TCM autógeno o alogénico.[13][Nivel de evidencia C2]
En una revisión retrospectiva de 84 pacientes con LACG negativo para ALK, se indicó un beneficio en la supervivencia con el TCM autógeno. Esta hipótesis requiere confirmación en un ensayo prospectivo aleatorizado.[14][Nivel de evidencia C3]
En un estudio retrospectivo, se incluyeron 182 pacientes con LACG en recaída o resistente al tratamiento (23 % positivos para ALK, 21 % negativos para ALK y 56 % con estado para ALK desconocido) sometidos a TCM alogénico.[15]
La tasa de SSP a 5 años fue del 41 % (IC 95 %, 34–49 %) y la tasa de SG a 5 años fue del 41 % (IC 95 %, 49–64 %).[15][Nivel de evidencia C3]
En el análisis multivariante, la raza afroamericana (CRI, 2,7; IC 95 %, 1,6–4,8; P < 0,001) y la enfermedad resistente al tratamiento en el momento del TCM alogénico (CRI, 3,2; IC 95 %, 1,6–6,2; P < 0,001) fueron factores predictivos de SG inferior.
Aunque la positividad para ALK fue un factor pronóstico favorable, en este estudio los desenlaces después de un TCM alogénico no variaron significativamente a partir del estado de ALK.
El LACG durante la niñez por lo general se caracteriza por enfermedad sistémica y cutánea con tasas de respuesta elevadas y una SG buena cuando se usa quimioterapia combinada a base de doxorrubicina.[16] El crizotinib, un inhibidor de ALK, combinado con quimioterapia se ha administrado en pacientes pediátricos sin tratamiento previo, y el crizotinib se ha usado para controlar la enfermedad en pacientes pediátricos con recaída múltiple.[17,18] El crizotinib se relaciona con un riesgo alto (alrededor del 25 %) de tromboembolia, en especial embolia pulmonar, por lo que se recomienda la profilaxis. No hay informes que respalden el uso de crizotinib en adultos.
Los pacientes con LACG asociado a implantes o prótesis de mama a veces evolucionan bien sin quimioterapia después de someterse a capsulectomía y extracción del implante si la enfermedad está confinada a la cápsula fibrosa y no hay masas ni linfadenopatía relacionadas.[19,20,21,22] La mayoría de los pacientes con LACG asociado a implantes de mama presentan una deleción característica en 20Q13.13 que quizás sea útil para el diagnóstico ya que ayuda a diferenciarlo del LACG cutáneo o sistémico.[23]
El LACG cutáneo primario es una entidad distinta que suele ser negativa para ALK y tiene una evolución clínica muy lenta (indolente o de grado bajo).
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Bai RY, Ouyang T, Miething C, et al.: Nucleophosmin-anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood 96 (13): 4319-27, 2000.
Hapgood G, Savage KJ: The biology and management of systemic anaplastic large cell lymphoma. Blood 126 (1): 17-25, 2015.
Gascoyne RD, Aoun P, Wu D, et al.: Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 93 (11): 3913-21, 1999.
Sibon D, Fournier M, Brière J, et al.: Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol 30 (32): 3939-46, 2012.
Horwitz S, O'Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019.
Horwitz S, O'Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022.
Younes A, Bartlett NL, Leonard JP, et al.: Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363 (19): 1812-21, 2010.
Pro B, Advani R, Brice P, et al.: Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 30 (18): 2190-6, 2012.
Prince HM, Kim YH, Horwitz SM, et al.: Brentuximab vedotin or physician's choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet 390 (10094): 555-566, 2017.
Pro B, Advani R, Brice P, et al.: Five-year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 130 (25): 2709-2717, 2017.
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012.
O'Connor OA, Horwitz S, Hamlin P, et al.: Phase II-I-II study of two different doses and schedules of pralatrexate, a high-affinity substrate for the reduced folate carrier, in patients with relapsed or refractory lymphoma reveals marked activity in T-cell malignancies. J Clin Oncol 27 (26): 4357-64, 2009.
Smith SM, Burns LJ, van Besien K, et al.: Hematopoietic cell transplantation for systemic mature T-cell non-Hodgkin lymphoma. J Clin Oncol 31 (25): 3100-9, 2013.
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022.
Furqan F, Ahn KW, Chen Y, et al.: Allogeneic haematopoietic cell transplant in patients with relapsed/refractory anaplastic large cell lymphoma. Br J Haematol 200 (1): 54-63, 2023.
Seidemann K, Tiemann M, Schrappe M, et al.: Short-pulse B-non-Hodgkin lymphoma-type chemotherapy is efficacious treatment for pediatric anaplastic large cell lymphoma: a report of the Berlin-Frankfurt-Münster Group Trial NHL-BFM 90. Blood 97 (12): 3699-706, 2001.
Lowe EJ, Reilly AF, Lim MS, et al.: Crizotinib in Combination With Chemotherapy for Pediatric Patients With ALK+ Anaplastic Large-Cell Lymphoma: The Results of Children's Oncology Group Trial ANHL12P1. J Clin Oncol 41 (11): 2043-2053, 2023.
Mossé YP, Voss SD, Lim MS, et al.: Targeting ALK With Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children's Oncology Group Study. J Clin Oncol 35 (28): 3215-3221, 2017.
Miranda RN, Aladily TN, Prince HM, et al.: Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol 32 (2): 114-20, 2014.
Clemens MW, Medeiros LJ, Butler CE, et al.: Complete Surgical Excision Is Essential for the Management of Patients With Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 34 (2): 160-8, 2016.
Mehta-Shah N, Clemens MW, Horwitz SM: How I treat breast implant-associated anaplastic large cell lymphoma. Blood 132 (18): 1889-1898, 2018.
Jaffe ES, Ashar BS, Clemens MW, et al.: Best Practices Guideline for the Pathologic Diagnosis of Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J Clin Oncol 38 (10): 1102-1111, 2020.
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020.
Tratamiento del linfoma angioinmunoblástico de células T
El linfoma angioinmunoblástico de células T (LACT) se llamaba antes linfadenopatía angioinmunoblástica con disproteinemia. Esta entidad, caracterizada por un reordenamiento génico clonal del receptor de células T, se trata como un linfoma difuso de células B grandes, con ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) (sin rituximab).[1,2,3,4] Los pacientes presentan linfadenopatía grave, fiebre, sudores nocturnos, pérdida de peso, exantema, prueba de Coombs positiva e hipergammaglobulinemia policlonal.[5] Las infecciones oportunistas son frecuentes debido a una inmunodeficiencia subyacente. En la mayoría de los pacientes afectados, se detectan genomas del virus de Epstein-Barr en las células B.[6] Para obtener más información sobre la pérdida de peso, consultar La nutrición en el tratamiento del cáncer y para obtener más información sobre el exantema, consultar Prurito.
La quimioterapia combinada a base de doxorrubicina, como el régimen CHOP, es la que más se usa para el LACT, al igual que para otros linfomas de crecimiento rápido (agresivos).[1,4] En los casos positivos para CD30, el tratamiento estándar propuesto es brentuximab vedotina combinado con ciclofosfamida, doxorrubicina y prednisona.[7][Nivel de evidencia C3] En un ensayo clínico, se incluyó a pacientes con LACT, en su mayoría con linfoma anaplásico de células grandes y no fue posible establecer un beneficio para este subgrupo más pequeño de LACT.[7,8][Nivel de evidencia C3] Para obtener más información, consultar la sección Tratamiento del linfoma anaplásico de células grandes.
En el International Peripheral T-Cell Lymphoma Project, en el que participaron 22 centros internacionales, se identificaron 243 pacientes con LACT; la tasa de supervivencia general a 5 años fue del 33 % y la tasa de supervivencia sin fracaso terapéutico fue del 18 %.[9] La quimioterapia mielosupresora y la radioterapia con apoyo de células madre periféricas autógenas o alogénicas se describieron en informes anecdóticos.[10,11,12,13,14,15][Nivel de evidencia C3] Hay informes anecdóticos de respuestas a ciclosporina,[16] pralatrexato,[17] bendamustina,[18] el inhibidor de la histona–desacetilasa romidepsina, y brentuximab vedotina (incluso si el linfoma tiene poca expresión de CD30 o ninguna).[19,20][Nivel de evidencia C3] De manera ocasional se han notificado remisiones espontáneas y respuestas prolongadas a los corticoesteroides.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Siegert W, Agthe A, Griesser H, et al.: Treatment of angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma using prednisone with or without the COPBLAM/IMVP-16 regimen. A multicenter study. Kiel Lymphoma Study Group. Ann Intern Med 117 (5): 364-70, 1992.
Jaffe ES: Angioimmunoblastic T-cell lymphoma: new insights, but the clinical challenge remains. Ann Oncol 6 (7): 631-2, 1995.
Siegert W, Nerl C, Agthe A, et al.: Angioimmunoblastic lymphadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol 6 (7): 659-64, 1995.
Bräuninger A, Spieker T, Willenbrock K, et al.: Survival and clonal expansion of mutating "forbidden" (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic t cell lymphoma. J Exp Med 194 (7): 927-40, 2001.
Horwitz S, O'Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019.
Horwitz S, O'Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022.
Federico M, Rudiger T, Bellei M, et al.: Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the international peripheral T-cell lymphoma project. J Clin Oncol 31 (2): 240-6, 2013.
Reimer P, Rüdiger T, Geissinger E, et al.: Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 27 (1): 106-13, 2009.
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
Kyriakou C, Canals C, Finke J, et al.: Allogeneic stem cell transplantation is able to induce long-term remissions in angioimmunoblastic T-cell lymphoma: a retrospective study from the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol 27 (24): 3951-8, 2009.
Park SI, Horwitz SM, Foss FM, et al.: The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer 125 (9): 1507-1517, 2019.
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022.
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020.
Advani R, Horwitz S, Zelenetz A, et al.: Angioimmunoblastic T cell lymphoma: treatment experience with cyclosporine. Leuk Lymphoma 48 (3): 521-5, 2007.
Amengual JE, Lichtenstein R, Lue J, et al.: A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131 (4): 397-407, 2018.
Damaj G, Gressin R, Bouabdallah K, et al.: Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol 31 (1): 104-10, 2013.
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012.
Fanale MA, Horwitz SM, Forero-Torres A, et al.: Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 131 (19): 2120-2124, 2018.
Tratamiento del linfoma periférico de células T sin otra indicación
Los pacientes con linfoma periférico de células T sin otra indicación (LPCT-SAI) presentan un linfoma difuso de células grandes o un linfoma difuso mixto que exhibe en la superficie celular un fenotipo postímico (periférico) de célula T que expresa CD4 o CD8, pero que no expresa ambos a la vez.[1] Los LPCT-SAI abarcan un grupo de linfomas ganglionares heterogéneos de células T que requerirán una división más detallada en el futuro.[2,3]
Pronóstico
La mayoría de los investigadores notifican tasas de respuesta y supervivencia más precarias para los pacientes con LPCT-SAI que para los pacientes con linfomas de crecimiento rápido de células B en estadios comparables.[3,4] La mayoría de los pacientes presentan múltiples factores de pronóstico adverso (es decir, edad avanzada, estadio IV, compromiso de múltiples sitios extraganglionares y concentraciones elevadas de lactato–deshidrogenasa), y tienen tasas bajas (<20 %) de supervivencia sin fracaso terapéutico y de supervivencia general (SG) a 5 años.[3,4] Al igual que con otros linfomas (por ejemplo, el linfoma difuso de células B grandes [LDCBG] o el linfoma folicular), la supervivencia sin complicaciones (SSC) a los 24 meses predice una tasa de SG a 5 años del 78 %.[5]
Abordajes terapéuticos
El tratamiento incluye quimioterapia combinada a base de doxorrubicina, como ciclofosfamida, doxorrubicina, vincristina y prednisona (CHOP) o CHOP con etopósido (CHOPE). Las dosis son las mismas que las que se usan para el LDCBG.[6] En los casos positivos para CD30, el tratamiento estándar propuesto es brentuximab vedotina combinado con ciclofosfamida, doxorrubicina y prednisona.[7][Nivel de evidencia C3] En un ensayo clínico, se incluyó a pacientes con LPCT-SAI en su mayoría con linfoma anaplásico de células grandes y no fue posible establecer un beneficio para este subgrupo más pequeño de LPCT-SAI.[7,8][Nivel de evidencia C3] Para obtener más información, consultar la sección Tratamiento del linfoma anaplásico de células grandes.
En pacientes con enfermedad en estadio temprano, los resultados anecdóticos de series retrospectivas son contradictorios en cuanto a la importancia de la radioterapia de consolidación después de la quimioterapia combinada.[9][Nivel de evidencia C3] En múltiples ensayos de fase II o retrospectivos, se administró terapia de consolidación con quimioterapia de dosis alta y trasplante de células madre (TCM) hematopoyéticas autógeno o alogénico a pacientes con LPCT en estadio avanzado después de la terapia de inducción. La evidencia que respalda este abordaje es anecdótica.[10,11,12,13,14,15,16,17][Nivel de evidencia C3]
En un ensayo prospectivo aleatorizado, se incluyeron 104 pacientes menores de 61 años con LPCT en estadio II, III o IV (excepto el linfoma anaplásico de células grandes positivo para ALK). Los pacientes recibieron un TCM autógeno o un TCM alogénico como terapia de consolidación después de la inducción con CHOPE seguido de DHAP (dexametasona, citarabina y cisplatino).[18][Nivel de evidencia C3]
Después de una mediana de seguimiento de 42 meses, la tasa de SSC a 3 años fue del 43 % para los pacientes que recibieron un TCM alogénico y del 38 % para los pacientes que recibieron un TCM autógeno.
La tasa de SG a 3 años fue del 57 % para los pacientes que recibieron un TCM alogénico y del 70 % para los pacientes que recibieron un TCM autógeno (P = no significativa).
Ninguno de los 21 pacientes que respondieron y que se sometieron a un TCM alogénico presentó recaída, mientras que el 36 % de los pacientes que se sometieron a un TCM autógeno sí presentaron recaída.
De 26 pacientes (31 %) que recibieron un TCM alogénico, 8 murieron por enfermedad de injerto contra huésped. Ninguno de los 41 pacientes sometidos a un TCM autógeno murió por toxicidad.
El aumento de la mortalidad relacionada con el trasplante anuló el beneficio del efecto injerto contra linfoma.
En un ensayo prospectivo de 109 pacientes evaluables con enfermedad recidivante, el tratamiento con pralatrexato produjo una tasa de respuesta del 30 % y una mediana de duración de la respuesta de 10 meses.[19,20][Nivel de evidencia C3]
Se observaron tasas de respuesta similares en 130 pacientes evaluables con enfermedad recidivante que recibieron romidepsina en un ensayo prospectivo.[20][Nivel de evidencia C3]
Se observaron respuestas anecdóticas con una combinación de pralatrexato y romidepsina,[21] bendamustina en monoterapia,[22] belinostat,[23] y brentuximab vedotina (incluso en linfomas con poca expresión de CD30 o ninguna).[24][Nivel de evidencia C3]
La incorporación de estos fármacos nuevos con quimioterapia CHOP está en evaluación clínica.[3,7]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Rüdiger T, Weisenburger DD, Anderson JR, et al.: Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin's Lymphoma Classification Project. Ann Oncol 13 (1): 140-9, 2002.
Weisenburger DD, Savage KJ, Harris NL, et al.: Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the International Peripheral T-cell Lymphoma Project. Blood 117 (12): 3402-8, 2011.
Sonnen R, Schmidt WP, Müller-Hermelink HK, et al.: The International Prognostic Index determines the outcome of patients with nodal mature T-cell lymphomas. Br J Haematol 129 (3): 366-72, 2005.
Maurer MJ, Ellin F, Srour L, et al.: International Assessment of Event-Free Survival at 24 Months and Subsequent Survival in Peripheral T-Cell Lymphoma. J Clin Oncol 35 (36): 4019-4026, 2017.
Carson KR, Horwitz SM, Pinter-Brown LC, et al.: A prospective cohort study of patients with peripheral T-cell lymphoma in the United States. Cancer 123 (7): 1174-1183, 2017.
Horwitz S, O'Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019.
Horwitz S, O'Connor OA, Pro B, et al.: The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 33 (3): 288-298, 2022.
Briski R, Feldman AL, Bailey NG, et al.: Survival in patients with limited-stage peripheral T-cell lymphomas. Leuk Lymphoma 56 (6): 1665-70, 2015.
Rodriguez J, Munsell M, Yazji S, et al.: Impact of high-dose chemotherapy on peripheral T-cell lymphomas. J Clin Oncol 19 (17): 3766-70, 2001.
Reimer P, Rüdiger T, Geissinger E, et al.: Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol 27 (1): 106-13, 2009.
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
Schmitz N, Lenz G, Stelljes M: Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood 132 (3): 245-253, 2018.
Park SI, Horwitz SM, Foss FM, et al.: The role of autologous stem cell transplantation in patients with nodal peripheral T-cell lymphomas in first complete remission: Report from COMPLETE, a prospective, multicenter cohort study. Cancer 125 (9): 1507-1517, 2019.
Brink M, Meeuwes FO, van der Poel MWM, et al.: Impact of etoposide and ASCT on survival among patients aged <65 years with stage II to IV PTCL: a population-based cohort study. Blood 140 (9): 1009-1019, 2022.
Los-de Vries GT, de Boer M, van Dijk E, et al.: Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 136 (25): 2927-2932, 2020.
Schmitz N, Truemper L, Bouabdallah K, et al.: A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood 137 (19): 2646-2656, 2021.
O'Connor OA, Pro B, Pinter-Brown L, et al.: Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29 (9): 1182-9, 2011.
Coiffier B, Pro B, Prince HM, et al.: Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol 30 (6): 631-6, 2012.
Amengual JE, Lichtenstein R, Lue J, et al.: A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 131 (4): 397-407, 2018.
Damaj G, Gressin R, Bouabdallah K, et al.: Results from a prospective, open-label, phase II trial of bendamustine in refractory or relapsed T-cell lymphomas: the BENTLY trial. J Clin Oncol 31 (1): 104-10, 2013.
O'Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015.
Fanale MA, Horwitz SM, Forero-Torres A, et al.: Five-year outcomes for frontline brentuximab vedotin with CHP for CD30-expressing peripheral T-cell lymphomas. Blood 131 (19): 2120-2124, 2018.
Tratamiento del linfoma extraganglionar de células T-NK
El linfoma extraganglionar de células T o de células citolíticas naturales (NK) (linfoma T/NK extraganglionar o linfoma extraganglionar NK/T) de tipo nasal es un linfoma de crecimiento rápido caracterizado por necrosis extensa y angioinvasión, que a menudo se presenta en sitios extraganglionares, en especial, en la región de la nariz o de los senos paranasales.[1] Otros sitios extraganglionares son el paladar, la tráquea, la piel y el tubo digestivo. Es posible que se presente un síndrome hemofagocítico; tradicionalmente, estos tumores se consideraban parte de un granuloma letal de línea media.[2] En la mayoría de los casos, se detectan genomas del virus de Epstein-Barr (VEB) en las células tumorales y en la inmunofenotipificación se observa un resultado positivo para CD56. Los casos con compromiso de la sangre y la médula se consideran leucemia de células NK. Es posible diferenciar una enteropatía benigna de células NK (negativa para VEB) de un linfoma de células T-NK mediante una biopsia endoscópica.[3]
Debido al aumento del riesgo de compromiso del sistema nervioso central y de recidiva local, se recomienda administrar radioterapia local antes del inicio de la quimioterapia o entre el segundo y el tercer ciclo, y para la profilaxis intratecal o la radioterapia craneal profiláctica.[4,5,6,7,8,9,10,11]
En una revisión retrospectiva se incluyeron 1273 pacientes con enfermedad en estadio temprano. Los pacientes se estratificaron en un grupo de riesgo bajo y un grupo de riesgo alto según el estadio, la edad, la concentración de lactato–deshidrogenasa, el estado funcional y la invasión del tumor primario.
Los pacientes de riesgo bajo evolucionaron mejor con radioterapia sola,[12] mientras que los pacientes de riesgo alto evolucionaron mejor con una estrategia de radioterapia combinada con quimioterapia.[10,13,14]
En una revisión retrospectiva, se incluyó 303 pacientes sin tratamiento previo, de un consorcio internacional, quienes recibieron quimioterapia sin antraciclinas.[15]
Las tasas de supervivencia general fueron idénticas (72−74 % a los 5 años) para los pacientes con enfermedad en estadio temprano que recibieron quimioterapia y radioterapia simultáneas o quimioterapia seguida de radioterapia.[15][Nivel de evidencia C3]
El uso de dosis más altas de radioterapia, superiores a 50 Gy, se relacionó con mejores desenlaces según informes anecdóticos.[10] La evolución muy maligna, la respuesta precaria y la supervivencia corta con el tratamiento estándar, en especial de los pacientes con enfermedad en estadio avanzado o presentación extranasal, llevó a algunos investigadores a recomendar la consolidación con un trasplante de células madre periféricas autógeno o alogénico.[11,16,17,18,19,20][Nivel de evidencia C3] En informes anecdóticos, los regímenes que contienen asparaginasa produjeron tasas de respuesta superiores al 50 % en pacientes con enfermedad recidivante, resistente al tratamiento o recién diagnosticada.[11,21,22,23,24][Nivel de evidencia C3] Debido a la falta de ensayos clínicos aleatorizados con más de 100 pacientes para este tipo poco frecuente de linfoma de células T, los regímenes que contienen pegaspargasa se han convertido en el tratamiento sistémico estándar. La pegaspargasa es una formulación menos tóxica de la asparaginasa que produce menos reacciones de hipersensibilidad y que tiene una semivida más larga.[25,26] El linfoma de células T-NK que se presenta solo en la piel tiene un pronóstico más favorable, en particular, en pacientes con coexpresión de CD30 y CD56.[27]
En un ensayo de fase II, se administró el ligando 1 de muerte programada (PD-L1) avelumab a 21 pacientes con enfermedad en recaída o resistente al tratamiento.[28]
La tasa de respuesta completa fue del 24 % y la tasa de respuesta general fue del 38 %. Las respuestas se correlacionaron con la expresión tumoral de PD-L1.[28][Nivel de evidencia C3]
El tratamiento con pembrolizumab, un anticuerpo contra la proteína de muerte programada 1 (PD-1), produjo respuestas similares en pacientes con enfermedad en recaída o resistente al tratamiento.[29][Nivel de evidencia C3]
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Tse E, Kwong YL: How I treat NK/T-cell lymphomas. Blood 121 (25): 4997-5005, 2013.
Mansoor A, Pittaluga S, Beck PL, et al.: NK-cell enteropathy: a benign NK-cell lymphoproliferative disease mimicking intestinal lymphoma: clinicopathologic features and follow-up in a unique case series. Blood 117 (5): 1447-52, 2011.
Li YX, Yao B, Jin J, et al.: Radiotherapy as primary treatment for stage IE and IIE nasal natural killer/T-cell lymphoma. J Clin Oncol 24 (1): 181-9, 2006.
Lee J, Suh C, Park YH, et al.: Extranodal natural killer T-cell lymphoma, nasal-type: a prognostic model from a retrospective multicenter study. J Clin Oncol 24 (4): 612-8, 2006.
Li CC, Tien HF, Tang JL, et al.: Treatment outcome and pattern of failure in 77 patients with sinonasal natural killer/T-cell or T-cell lymphoma. Cancer 100 (2): 366-75, 2004.
Yamaguchi M, Tobinai K, Oguchi M, et al.: Phase I/II study of concurrent chemoradiotherapy for localized nasal natural killer/T-cell lymphoma: Japan Clinical Oncology Group Study JCOG0211. J Clin Oncol 27 (33): 5594-600, 2009.
Kim SJ, Kim K, Kim BS, et al.: Phase II trial of concurrent radiation and weekly cisplatin followed by VIPD chemotherapy in newly diagnosed, stage IE to IIE, nasal, extranodal NK/T-Cell Lymphoma: Consortium for Improving Survival of Lymphoma study. J Clin Oncol 27 (35): 6027-32, 2009.
Li YX, Fang H, Liu QF, et al.: Clinical features and treatment outcome of nasal-type NK/T-cell lymphoma of Waldeyer ring. Blood 112 (8): 3057-64, 2008.
Vargo JA, Patel A, Glaser SM, et al.: The impact of the omission or inadequate dosing of radiotherapy in extranodal natural killer T-cell lymphoma, nasal type, in the United States. Cancer 123 (16): 3176-3185, 2017.
Yamaguchi M, Suzuki R, Oguchi M: Advances in the treatment of extranodal NK/T-cell lymphoma, nasal type. Blood 131 (23): 2528-2540, 2018.
Yang Y, Cao JZ, Lan SM, et al.: Association of Improved Locoregional Control With Prolonged Survival in Early-Stage Extranodal Nasal-Type Natural Killer/T-Cell Lymphoma. JAMA Oncol 3 (1): 83-91, 2017.
Yang Y, Zhu Y, Cao JZ, et al.: Risk-adapted therapy for early-stage extranodal nasal-type NK/T-cell lymphoma: analysis from a multicenter study. Blood 126 (12): 1424-32; quiz 1517, 2015.
Yamaguchi M, Suzuki R, Oguchi M, et al.: Treatments and Outcomes of Patients With Extranodal Natural Killer/T-Cell Lymphoma Diagnosed Between 2000 and 2013: A Cooperative Study in Japan. J Clin Oncol 35 (1): 32-39, 2017.
Kwong YL, Kim SJ, Tse E, et al.: Sequential chemotherapy/radiotherapy was comparable with concurrent chemoradiotherapy for stage I/II NK/T-cell lymphoma. Ann Oncol 29 (1): 256-263, 2018.
Liang R, Todd D, Chan TK, et al.: Treatment outcome and prognostic factors for primary nasal lymphoma. J Clin Oncol 13 (3): 666-70, 1995.
Cheung MM, Chan JK, Lau WH, et al.: Primary non-Hodgkin's lymphoma of the nose and nasopharynx: clinical features, tumor immunophenotype, and treatment outcome in 113 patients. J Clin Oncol 16 (1): 70-7, 1998.
Hausdorff J, Davis E, Long G, et al.: Non-Hodgkin's lymphoma of the paranasal sinuses: clinical and pathological features, and response to combined-modality therapy. Cancer J Sci Am 3 (5): 303-11, 1997 Sep-Oct.
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
Au WY, Weisenburger DD, Intragumtornchai T, et al.: Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood 113 (17): 3931-7, 2009.
Jaccard A, Gachard N, Marin B, et al.: Efficacy of L-asparaginase with methotrexate and dexamethasone (AspaMetDex regimen) in patients with refractory or relapsing extranodal NK/T-cell lymphoma, a phase 2 study. Blood 117 (6): 1834-9, 2011.
Yamaguchi M, Kwong YL, Kim WS, et al.: Phase II study of SMILE chemotherapy for newly diagnosed stage IV, relapsed, or refractory extranodal natural killer (NK)/T-cell lymphoma, nasal type: the NK-Cell Tumor Study Group study. J Clin Oncol 29 (33): 4410-6, 2011.
Li JW, Li YJ, Zhong MZ, et al.: Efficacy and tolerance of GELOXD/P-GEMOXD in newly diagnosed nasal-type extranodal NK/T-cell lymphoma: A multicenter retrospective study. Eur J Haematol 100 (3): 247-256, 2018.
Wei L, Wang L, Cong J, et al.: SVILE regimen, a combination of dexamethasone, vindesine, ifosfamide, pegaspargase, and etoposide, for treating relapsed/refractory extranodal natural killer/T-cell lymphoma, nasal type. Leuk Res 96: 106422, 2020.
Liang R, Gao GX, Chen JP, et al.: A phase 2 study of methotrexate, etoposide, dexamethasone, and pegaspargase chemotherapy for newly diagnosed, relapsed, or refractory extranodal natural killer/T-cell lymphoma, nasal type: a multicenter trial in Northwest China. Hematol Oncol 35 (4): 619-629, 2017.
Wang X, Zhang L, Liu X, et al.: Efficacy and Safety of a Pegasparaginase-Based Chemotherapy Regimen vs an L-asparaginase-Based Chemotherapy Regimen for Newly Diagnosed Advanced Extranodal Natural Killer/T-Cell Lymphoma: A Randomized Clinical Trial. JAMA Oncol 8 (7): 1035-1041, 2022.
Mraz-Gernhard S, Natkunam Y, Hoppe RT, et al.: Natural killer/natural killer-like T-cell lymphoma, CD56+, presenting in the skin: an increasingly recognized entity with an aggressive course. J Clin Oncol 19 (8): 2179-88, 2001.
Kim SJ, Lim JQ, Laurensia Y, et al.: Avelumab for the treatment of relapsed or refractory extranodal NK/T-cell lymphoma: an open-label phase 2 study. Blood 136 (24): 2754-2763, 2020.
Kwong YL, Chan TSY, Tan D, et al.: PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 129 (17): 2437-2442, 2017.
Tratamiento del linfoma de células T asociado a enteropatía
El linfoma de células T asociado a enteropatía afecta el intestino delgado de pacientes con enfermedad celíaca (celiaquía o enteropatía por gluten).[1,2,3,4] Debido a que la alimentación sin gluten previene el linfoma en los pacientes con enfermedad celíaca en la niñez, rara vez estos pacientes presentan linfoma. El diagnóstico de enfermedad celíaca se suele establecer por la presencia de atrofia vellosa del intestino resecado. A menudo se necesita una cirugía para establecer el diagnóstico y evitar la perforación intestinal durante el tratamiento.
El tratamiento es quimioterapia combinada a base de doxorrubicina, pero las tasas de recaída son más altas que para el linfoma difuso de células grandes en estadio comparable.[2,4,5] Las complicaciones del tratamiento son hemorragia gastrointestinal, perforación del intestino delgado y fístula enterocólica y a menudo los pacientes necesitan nutrición parenteral. Para obtener más información sobre la nutrición parenteral, consultar La nutrición en el tratamiento del cáncer. En el momento de la recaída se observan perforaciones intestinales multifocales y compromiso de vísceras abdominales. La terapia de dosis altas con rescate de células madre hematopoyéticas se ha usado en el momento de la primera remisión o en la recaída.[2,6,7][Nivel de evidencia C3] La evidencia que respalda este abordaje es anecdótica.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Egan LJ, Walsh SV, Stevens FM, et al.: Celiac-associated lymphoma. A single institution experience of 30 cases in the combination chemotherapy era. J Clin Gastroenterol 21 (2): 123-9, 1995.
Gale J, Simmonds PD, Mead GM, et al.: Enteropathy-type intestinal T-cell lymphoma: clinical features and treatment of 31 patients in a single center. J Clin Oncol 18 (4): 795-803, 2000.
Di Sabatino A, Biagi F, Gobbi PG, et al.: How I treat enteropathy-associated T-cell lymphoma. Blood 119 (11): 2458-68, 2012.
Daum S, Ullrich R, Heise W, et al.: Intestinal non-Hodgkin's lymphoma: a multicenter prospective clinical study from the German Study Group on Intestinal non-Hodgkin's Lymphoma. J Clin Oncol 21 (14): 2740-6, 2003.
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
Sieniawski M, Angamuthu N, Boyd K, et al.: Evaluation of enteropathy-associated T-cell lymphoma comparing standard therapies with a novel regimen including autologous stem cell transplantation. Blood 115 (18): 3664-70, 2010.
Tratamiento del linfoma hepatoesplénico de células T
El linfoma hepatoesplénico de células T es un tipo poco común de linfoma periférico de células T que se presenta en su mayoría en hombres jóvenes. El linfoma hepatoesplénico de células T está localizado en los sinusoides hepáticos y esplénicos, y en la superficie celular expresa el receptor de células T γ-δ.[1,2,3] Este linfoma tiene un pronóstico muy precario y una evolución clínica muy maligna. El linfoma hepatoesplénico de células T se trata con quimioterapia de inducción y consolidación con trasplante de células madre (TCM).[3,4]
En un metanálisis de 118 pacientes con linfoma hepatoesplénico de células T, se comparó el régimen CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona) con regímenes de inducción similares a CHOP.[5]
Los regímenes diferentes a CHOP (que contienen citarabina, etopósido o derivados del platino) se relacionaron con mejores desenlaces, incluso una tasa de respuesta general del 82 % versus el 52 % (P = 0,006) y una mediana de supervivencia general (SG) de 36,5 versus 18 meses (P = 0,00014).
La consolidación con TCM alogénico se relacionó con una mediana de SG mejor de 33 meses versus 27 meses (P = 0,016) para el TCM autógeno.[5][Nivel de evidencia C3]
El uso de ICE (ifosfamida, carboplatino y etopósido) o IVAC (ifosfamida, etopósido y dosis altas de citarabina) produjo mejores respuestas cuando se comparó con CHOP en otros estudios más pequeños.[6][Nivel de evidencia D] Debido a las respuestas inadecuadas a los regímenes CHOP o similares a estos, muchos profesionales clínicos usan la quimioterapia ICE como terapia de inducción de primera línea, seguida de un TCM alogénico de consolidación en los pacientes que logran una primera remisión. Sin embargo, la eficacia de este abordaje es indeterminada.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Belhadj K, Reyes F, Farcet JP, et al.: Hepatosplenic gammadelta T-cell lymphoma is a rare clinicopathologic entity with poor outcome: report on a series of 21 patients. Blood 102 (13): 4261-9, 2003.
Chanan-Khan A, Islam T, Alam A, et al.: Long-term survival with allogeneic stem cell transplant and donor lymphocyte infusion following salvage therapy with anti-CD52 monoclonal antibody (Campath) in a patient with alpha/beta hepatosplenic T-cell non-Hodgkin's lymphoma. Leuk Lymphoma 45 (8): 1673-5, 2004.
Pro B, Allen P, Behdad A: Hepatosplenic T-cell lymphoma: a rare but challenging entity. Blood 136 (18): 2018-2026, 2020.
Le Gouill S, Milpied N, Buzyn A, et al.: Graft-versus-lymphoma effect for aggressive T-cell lymphomas in adults: a study by the Société Francaise de Greffe de Moëlle et de Thérapie Cellulaire. J Clin Oncol 26 (14): 2264-71, 2008.
Klebaner D, Koura D, Tzachanis D, et al.: Intensive Induction Therapy Compared With CHOP for Hepatosplenic T-cell Lymphoma. Clin Lymphoma Myeloma Leuk 20 (7): 431-437.e2, 2020.
Voss MH, Lunning MA, Maragulia JC, et al.: Intensive induction chemotherapy followed by early high-dose therapy and hematopoietic stem cell transplantation results in improved outcome for patients with hepatosplenic T-cell lymphoma: a single institution experience. Clin Lymphoma Myeloma Leuk 13 (1): 8-14, 2013.
Tratamiento de la leucemia o linfoma de células T en adultos
La leucemia o linfoma de células T (LLCTA) en adultos se debe a una infección por un retrovirus llamado virus linfotrópico humano de células T de tipo 1 (HTLV1) y, con frecuencia, se relaciona con linfadenopatía, hipercalcemia, células leucémicas circulantes, compromiso óseo y cutáneo, hepatoesplenomegalia, progresión rápida y respuesta precaria a la quimioterapia combinada.[1,2] Esta enfermedad se divide en cuatro subtipos clínicos:[3,4]
Tipo agudo (evolución muy maligna, es decir, de crecimiento rápido o agresiva, con leucemia y compromiso extraganglionar o ganglionar, o sin este).
Tipo linfoma (evolución muy maligna, es decir de crecimiento rápido o agresivo, con linfadenopatía y sin leucemia).
Tipo crónico (evolución de escasa malignidad, es decir de crecimiento lento o indolente, con leucemia y linfadenopatía).
Tipo quiescente (evolución de escasa malignidad, es decir de crecimiento lento o indolente, solo con leucemia).
La enfermedad de tipo aguda y de tipo linfoma responden de manera precaria a la quimioterapia combinada y al trasplante de células madre (TCM) alogénico, y tienen una mediana de supervivencia general (SG) inferior a 1 año.[5,6,7] Menos del 10 % de los 807 pacientes que recibieron terapia combinada estaban vivos después de 4 años.[7] Hay informes anecdóticos de remisiones duraderas después de un TCM alogénico e incluso tras una infusión de linfocitos de un donante para recaídas posteriores al trasplante.[8][Nivel de evidencia C3] Entre 815 pacientes sometidos a TCM alogénico en dos revisiones retrospectivas, las tasas de SG a 3 años fueron del 36 % y el 26 %.[9,10][Nivel de evidencia C1]
La combinación de zidovudina e interferón α es eficaz contra la LLCTA, incluso en pacientes con fracaso del tratamiento citotóxico previo. En la mayoría de los pacientes tratados con esta combinación se observan remisiones duraderas, que no se observan en pacientes con el subtipo linfoma.[11,12,13,14,15] En un estudio multicéntrico de fase II de 26 pacientes con recaída, el 42 % respondió a la lenalidomida (incluidas 4 respuestas completas).[16][Nivel de evidencia C3] La progresión local sintomática de todos los subtipos responde bien a la radioterapia paliativa.[17] En el entorno de recaída, se observó una tasa de respuesta general superior al 50 % con mogamulizumab, un anticuerpo monoclonal humanizado contra el receptor de quimiocina CC 4 (CCR4).[18][Nivel de evidencia C3] En los casos positivos para CD30, el tratamiento estándar es brentuximab vedotina combinado con ciclofosfamida, doxorrubicina y prednisona.[19] En un ensayo clínico, se incluyeron pacientes con LLCTA en su mayoría pacientes con linfoma anaplásico de células grandes; no fue posible establecer un beneficio para este subgrupo más pequeño de LLCTA.[19][Nivel de evidencia C3] Para obtener más información, consultar la sección Tratamiento del linfoma anaplásico de células grandes.
Ensayos clínicos en curso
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Höllsberg P, Hafler DA: Seminars in medicine of the Beth Israel Hospital, Boston. Pathogenesis of diseases induced by human lymphotropic virus type I infection. N Engl J Med 328 (16): 1173-82, 1993.
Foss FM, Aquino SL, Ferry JA: Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 10-2003. A 72-year-old man with rapidly progressive leukemia, rash, and multiorgan failure. N Engl J Med 348 (13): 1267-75, 2003.
Shimoyama M: Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984-87). Br J Haematol 79 (3): 428-37, 1991.
Takasaki Y, Iwanaga M, Imaizumi Y, et al.: Long-term study of indolent adult T-cell leukemia-lymphoma. Blood 115 (22): 4337-43, 2010.
Yamada Y, Tomonaga M, Fukuda H, et al.: A new G-CSF-supported combination chemotherapy, LSG15, for adult T-cell leukaemia-lymphoma: Japan Clinical Oncology Group Study 9303. Br J Haematol 113 (2): 375-82, 2001.
Fukushima T, Miyazaki Y, Honda S, et al.: Allogeneic hematopoietic stem cell transplantation provides sustained long-term survival for patients with adult T-cell leukemia/lymphoma. Leukemia 19 (5): 829-34, 2005.
Katsuya H, Yamanaka T, Ishitsuka K, et al.: Prognostic index for acute- and lymphoma-type adult T-cell leukemia/lymphoma. J Clin Oncol 30 (14): 1635-40, 2012.
Itonaga H, Tsushima H, Taguchi J, et al.: Treatment of relapsed adult T-cell leukemia/lymphoma after allogeneic hematopoietic stem cell transplantation: the Nagasaki Transplant Group experience. Blood 121 (1): 219-25, 2013.
Ishida T, Hishizawa M, Kato K, et al.: Allogeneic hematopoietic stem cell transplantation for adult T-cell leukemia-lymphoma with special emphasis on preconditioning regimen: a nationwide retrospective study. Blood 120 (8): 1734-41, 2012.
Katsuya H, Ishitsuka K, Utsunomiya A, et al.: Treatment and survival among 1594 patients with ATL. Blood 126 (24): 2570-7, 2015.
Gill PS, Harrington W, Kaplan MH, et al.: Treatment of adult T-cell leukemia-lymphoma with a combination of interferon alfa and zidovudine. N Engl J Med 332 (26): 1744-8, 1995.
Matutes E, Taylor GP, Cavenagh J, et al.: Interferon alpha and zidovudine therapy in adult T-cell leukaemia lymphoma: response and outcome in 15 patients. Br J Haematol 113 (3): 779-84, 2001.
Hermine O, Allard I, Lévy V, et al.: A prospective phase II clinical trial with the use of zidovudine and interferon-alpha in the acute and lymphoma forms of adult T-cell leukemia/lymphoma. Hematol J 3 (6): 276-82, 2002.
Bazarbachi A, Plumelle Y, Carlos Ramos J, et al.: Meta-analysis on the use of zidovudine and interferon-alfa in adult T-cell leukemia/lymphoma showing improved survival in the leukemic subtypes. J Clin Oncol 28 (27): 4177-83, 2010.
Bazarbachi A, Suarez F, Fields P, et al.: How I treat adult T-cell leukemia/lymphoma. Blood 118 (7): 1736-45, 2011.
Ishida T, Fujiwara H, Nosaka K, et al.: Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATLL-002. J Clin Oncol 34 (34): 4086-4093, 2016.
Simone CB, Morris JC, Stewart DM, et al.: Radiation therapy for the management of patients with HTLV-1-associated adult T-cell leukemia/lymphoma. Blood 120 (9): 1816-9, 2012.
Ureshino H, Kamachi K, Kimura S: Mogamulizumab for the Treatment of Adult T-cell Leukemia/Lymphoma. Clin Lymphoma Myeloma Leuk 19 (6): 326-331, 2019.
Horwitz S, O'Connor OA, Pro B, et al.: Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): a global, double-blind, randomised, phase 3 trial. Lancet 393 (10168): 229-240, 2019.
Tratamiento del linfoma periférico de células T en recaída o resistente al tratamiento
Opciones de tratamiento del linfoma periférico de células T en recaída o resistente al tratamiento
Las opciones de tratamiento del linfoma periférico de células T en recaída o resistente al tratamiento son las siguientes:
Realizar una búsqueda avanzada en inglés de los ensayos clínicos sobre cáncer auspiciados por el NCI que ahora aceptan pacientes. La búsqueda se puede simplificar por ubicación del ensayo, tipo de tratamiento, nombre del fármaco y otros criterios. También se dispone de información general sobre los ensayos clínicos.
Referencias:
Jerkeman M, Leppä S, Kvaløy S, et al.: ICE (ifosfamide, carboplatin, etoposide) as second-line chemotherapy in relapsed or primary progressive aggressive lymphoma--the Nordic Lymphoma Group experience. Eur J Haematol 73 (3): 179-82, 2004.
Zelenetz AD, Hamlin P, Kewalramani T, et al.: Ifosfamide, carboplatin, etoposide (ICE)-based second-line chemotherapy for the management of relapsed and refractory aggressive non-Hodgkin's lymphoma. Ann Oncol 14 (Suppl 1): i5-10, 2003.
Shen QD, Wang L, Zhu HY, et al.: Gemcitabine, oxaliplatin and dexamethasone (GemDOx) as salvage therapy for relapsed or refractory diffuse large B-cell lymphoma and peripheral T-cell lymphoma. J Cancer 12 (1): 163-169, 2021.
Schmitz N, Truemper L, Bouabdallah K, et al.: A randomized phase 3 trial of autologous vs allogeneic transplantation as part of first-line therapy in poor-risk peripheral T-NHL. Blood 137 (19): 2646-2656, 2021.
Velasquez WS, McLaughlin P, Tucker S, et al.: ESHAP--an effective chemotherapy regimen in refractory and relapsing lymphoma: a 4-year follow-up study. J Clin Oncol 12 (6): 1169-76, 1994.
Mercadal S, Briones J, Xicoy B, et al.: Intensive chemotherapy (high-dose CHOP/ESHAP regimen) followed by autologous stem-cell transplantation in previously untreated patients with peripheral T-cell lymphoma. Ann Oncol 19 (5): 958-63, 2008.
Hapgood G, Stone JM, Zannino D, et al.: A phase II study of a modified hyper-CVAD frontline therapy for patients with adverse risk diffuse large B-cell and peripheral T-cell non-Hodgkin lymphoma. Leuk Lymphoma 60 (4): 904-911, 2019.
Iyer SP, Foss FF: Romidepsin for the Treatment of Peripheral T-Cell Lymphoma. Oncologist 20 (9): 1084-91, 2015.
Campbell P, Thomas CM: Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J Oncol Pharm Pract 23 (2): 143-147, 2017.
O'Connor OA, Horwitz S, Masszi T, et al.: Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J Clin Oncol 33 (23): 2492-9, 2015.
O'Connor OA, Pro B, Pinter-Brown L, et al.: Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol 29 (9): 1182-9, 2011.
Schmitz N, Lenz G, Stelljes M: Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood 132 (3): 245-253, 2018.
Chen AI, McMillan A, Negrin RS, et al.: Long-term results of autologous hematopoietic cell transplantation for peripheral T cell lymphoma: the Stanford experience. Biol Blood Marrow Transplant 14 (7): 741-7, 2008.
Hamadani M, Ngoya M, Sureda A, et al.: Outcome of allogeneic transplantation for mature T-cell lymphomas: impact of donor source and disease characteristics. Blood Adv 6 (3): 920-930, 2022.
Mehta-Shah N, Kommalapati A, Teja S: Successful treatment of mature T-cell lymphoma with allogeneic stem cell transplantation: the largest multicenter retrospective analysis. [Abstract] Blood 136 (Suppl 1): A-624, 35-36, 2020.
Sterling CH, Hughes MS, Tsai HL, et al.: Allogeneic Blood or Marrow Transplantation with Post-Transplantation Cyclophosphamide for Peripheral T Cell Lymphoma: The Importance of Graft Source. Transplant Cell Ther 29 (4): 267.e1-267.e5, 2023.
Actualizaciones más recientes a este resumen (07 / 11 / 2024)
Los resúmenes del PDQ con información sobre el cáncer se revisan con regularidad y se actualizan a medida que se obtiene nueva información. Esta sección describe los cambios más recientes introducidos en este resumen a partir de la fecha arriba indicada.
Se incorporaron cambios editoriales en este resumen.
El Consejo editorial del PDQ sobre el tratamiento para adultos es responsable de la redacción y actualización de este resumen y mantiene independencia editorial respecto del NCI. El resumen refleja una revisión independiente de la bibliografía médica y no representa las políticas del NCI ni de los NIH. Para obtener más información sobre las políticas relativas a los resúmenes y la función de los consejos editoriales del PDQ responsables de su actualización, consultar Información sobre este resumen del PDQ e Información del PDQ® sobre el cáncer dirigida a profesionales de la salud.
Información sobre este resumen del PDQ
Propósito de este resumen
Este resumen de información del PDQ sobre el cáncer dirigido a profesionales de la salud proporciona información integral revisada por expertos y basada en la evidencia sobre el tratamiento del linfoma no Hodgkin periférico de células T en adultos. El objetivo es servir como fuente de información y ayuda para los profesionales clínicos durante la atención de pacientes. No ofrece pautas ni recomendaciones formales para tomar decisiones relacionadas con la atención sanitaria.
Revisores y actualizaciones
El consejo editorial del PDQ sobre el tratamiento para adultos, que mantiene independencia editorial respecto del Instituto Nacional del Cáncer (NCI), revisa este resumen de manera periódica y, en caso necesario, lo actualiza. Este resumen es el resultado de una revisión bibliográfica independiente y no constituye una declaración de política del NCI ni de los Institutos Nacionales de la Salud (NIH).
Cada mes, los integrantes de este consejo revisan los artículos publicados recientemente para determinar lo siguiente:
Si el artículo se debe analizar en una reunión del consejo.
Si conviene añadir texto acerca del artículo.
Si se debe reemplazar o actualizar un artículo que ya se citó.
Los cambios en los resúmenes se deciden mediante consenso de los integrantes del consejo después de evaluar la solidez de la evidencia de los artículos publicados y determinar la forma de incorporar el artículo en el resumen.
Los revisores principales del sumario sobre Tratamiento del linfoma no Hodgkin periférico de células T son:
Eric J. Seifter, MD (Johns Hopkins University)
Cole H. Sterling, MD (Johns Hopkins University)
Cualquier comentario o pregunta sobre el contenido de este resumen se debe enviar al Servicio de Información de Cáncer del Instituto Nacional del Cáncer. Por favor, no enviar preguntas o comentarios directamente a los integrantes del consejo, ya que no responderán consultas de manera individual.
Niveles de evidencia
Algunas de las referencias bibliográficas de este resumen se acompañan del nivel de evidencia. El propósito de esto es ayudar al lector a evaluar la solidez de la evidencia que respalda el uso de ciertas intervenciones o abordajes. El consejo editorial del PDQ sobre el tratamiento para adultos emplea un sistema de jerarquización formal para asignar los niveles de evidencia científica.
Permisos para el uso de este resumen
PDQ (Physician Data Query) es una marca registrada. Se autoriza el uso del texto de los documentos del PDQ; sin embargo, no se podrá identificar como un resumen de información sobre cáncer del PDQ del NCI, salvo que el resumen se reproduzca en su totalidad y se actualice de manera periódica. Por otra parte, se permitirá que un autor escriba una oración como "En el resumen del PDQ del NCI de información sobre la prevención del cáncer de mama se describen, de manera concisa, los siguientes riesgos: [incluir fragmento del resumen]".
Se sugiere citar la referencia bibliográfica de este resumen del PDQ de la siguiente forma:
Las imágenes en este resumen se reproducen con autorización del autor, el artista o la editorial para uso exclusivo en los resúmenes del PDQ. La utilización de las imágenes fuera del PDQ requiere la autorización del propietario, que el Instituto Nacional del Cáncer no puede otorgar. Para obtener más información sobre el uso de las ilustraciones de este resumen o de otras imágenes relacionadas con el cáncer, consultar Visuals Online, una colección de más de 2000 imágenes científicas.
Cláusula sobre el descargo de responsabilidad
Según la solidez de la evidencia, las opciones de tratamiento se clasifican como "estándar" o "en evaluación clínica". Estas clasificaciones no se deben utilizar para justificar decisiones sobre reembolsos de seguros. Para obtener más información sobre la cobertura de seguros, consultar la página Manejo de la atención del cáncer en Cancer.gov/espanol.
Healthwise, Healthwise para cada decisión de la salud, y el logo de Healthwise son marcas de fábrica de Ignite Healthwise, LLC.
La Enciclopedia de salud contiene información general de salud. No todos los tratamientos o servicios descritos son beneficios cubiertos para los miembros de Kaiser Permanente ni se ofrecen como servicios de Kaiser Permanente. Para obtener una lista de beneficios cubiertos, consulte su Evidencia de cobertura o Descripción resumida del plan. Para los tratamientos recomendados, consulte con su proveedor de atención médica.